Building and evaluation of a PBPK model for fluvoxamine in healthy adults¶
| Version | 2.0-OSP12.3 |
|---|---|
| based on Model Snapshot and Evaluation Plan | https://github.com/Open-Systems-Pharmacology/Fluvoxamine-Model/releases/tag/v2.0 |
| OSP Version | 12.3 |
| Qualification Framework Version | 3.7 |
This evaluation report and the corresponding PK-Sim project file are filed at:
https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/
Table of Contents¶
- 1 Introduction
- 2 Methods
- 2.1 Modeling Strategy
- 2.2 Data
- 2.3 Model Parameters and Assumptions
- 3 Results and Discussion
- 3.1 Final input parameters
- 3.2 Diagnostics Plots
- 3.3 Concentration-Time Profiles
- 4 Conclusion
- 5 References
1 Introduction¶
Fluvoxamine is a selective serotonin reuptake inhibitor used to treat major depression and obsessive compulsive disorder (Perucca 1994, ANI Pharmaceuticals Inc. 2008) . Recommended doses are 50 to 300 mg once daily. The pharmacokinetics of orally administered single doses are linear. Following multiple oral administration, the pharmacokinetics at steady-state become non-linear, due to saturable Michaelis-Menten kinetics of the metabolic pathways (Spigset 1998). Metabolism of fluvoxamine includes hydroxylation via CYP1A2 and O-demethylation via the very polymorphic CYP2D6 (Miura 2007, Spigset 2001). Following oral administration fluvoxamine is excreted via the urine as metabolites (DeBree 1983). The U.S. Food and Drug Administration (FDA) recommends fluvoxamine as strong clinical CYP1A2 and CYP2C19 index inhibitor to evaluate the impact of CYP1A2/CYP2C19 inhibition on CYP1A2/CYP2C19 substrates (FDA 2017). Furthermore, the FDA lists fluvoxamine as moderate CYP3A4 inhibitor.
The aim of this project was to develop a PBPK model of fluvoxamine, mechanistically describing its metabolism by CYP1A2 and CYP2D6 and its inhibitory effect on CYP1A2 and CYP3A4, that can be used for drug-drug interaction (DDI) predictions.
The presented model was developed and evaluated by Britz et al. (Britz 2019)
2 Methods¶
2.1 Modeling Strategy¶
The general workflow for building an adult PBPK model has been described by Kuepfer et al. (Kuepfer 2016). Relevant information on the anthropometry (height, weight) was gathered from the respective clinical study, if reported. Information on physiological parameters (e.g. blood flows, organ volumes, hematocrit) in adults was gathered from the literature and has been incorporated in PK-Sim® as described previously (Willmann 2007). The applied activity and variability of plasma proteins and active processes that are integrated into PK-Sim® are described in the publicly available 'PK-Sim® Ontogeny Database Version 7.3' (PK-Sim Ontogeny Database Version 7.3).
The PBPK model was built based on healthy individuals, using the reported mean values for age, weight, height, and genetic background for each study protocol. If no information on these parameters could be found, a healthy male European individual, 30 years of age, with a body weight of 73 kg and a height of 176 cm was used. To model the specific metabolic clearance, CYP1A2 and CYP2D6 were implemented in accordance with literature, using the PK-Sim expression database RT-PCR profiles (Meyer 2012) to define their relative expression in the different organs of the body. Glomerular filtration and enterohepatic cycling were enabled, as they are involved in fluvoxamine excretion.
Unknown parameters (see below) were identified using the Parameter Identification module provided in PK-Sim®.
The model was then verified by simulating:
- single and multiple dose studies
- the effect of smoking on CYP1A2 metabolism of fluvoxamine
- plasma levels of fluvoxamine in CYP2D6 extensive (EM) and poor metabolizers (PM).
Details about input data (physicochemical, in vitro and clinical) can be found in Section 2.2.
Details about the structural model and its parameters can be found in Section 2.3.
2.2 Data¶
2.2.1 In vitro / physico-chemical Data¶
A literature search was performed to collect available information on physicochemical properties of fluvoxamine. The obtained information from literature is summarized in the table below.
| Parameter | Unit | Value | Source | Description |
|---|---|---|---|---|
| MW | g/mol | 318.34 | Drugbank | Molecular weight |
| pKa | 9.40 (base) | Hallifax 2007 | Acid dissociation constant | |
| Solubility (pH) | mg/mL | 14.66 (7.0) | MSDS | Solubility |
| logP | 2.80 | Drugbank (predicted by ChemAxon) | Partition coefficient between octanol and water | |
| 2.89 | Drugbank (predicted by ALOGPS) | Partition coefficient between octanol and water | ||
| 3.20 | Drugbank (experimentally measured) | Partition coefficient between octanol and water | ||
| fu | 0.13 ± 0.01a | Yao 2001 | Fraction unbound in plasma | |
| 0.14 ± 0.02a | Yao 2001 | Fraction unbound in plasma | ||
| 0.23 | Claassen 1983 | Fraction unbound in plasma | ||
| fu,mic | 0.20 ± 0.05a | Yao 2001 | Fraction unbound in human liver microsomes at a protein concentration of 1 mg/mL | |
| 0.31 ± 0.03a | Yao 2001 | Fraction unbound in human liver microsomes at a protein concentration of 0.5 mg/mL | ||
| 0.70 ± 0.03a | Yao 2001 | Fraction unbound in supersomes at a protein concentration of 0.3 mg/mL | ||
| CYP2D6 Km | µmol/L | 76.30 | Miura 2007 | Michaelis-Menten constant |
| CYP2D6 kcat | 1/min | 0 | Crews 2014 | The number of substrate molecule each enzyme site converts to product per unit time, and in which the enzyme is working at maximum efficiency |
| CYP1A2 Ki | µmol/L | 0.011 | Karjalainen 2008 | Competitive inhibition constant of the competitive inhibition model measured in human liver microsomes |
| CYP1A2 Ki,u | nmol/L | 35 | Yao 2001 | Unbound competitive inhibition constant of the mixed inhibition model measured in human liver microsomes at a protein concentration of 1 mg/mL |
| nmol/L | 36 | Yao 2001 | Competitive inhibition constant of the mixed inhibition model measured in human liver microsomes at a protein concentration of 0.5 mg/mL | |
| nmol/L | 36 | Yao 2001 | Competitive inhibition constant of the mixed inhibition model measured in supersomes at a protein concentration of 0.3 mg/mL | |
| CYP3A4 Ki | µmol/L | 1.60 | Olesen 2000 | Competitive inhibition constant of the competitive inhibition model measured in human liver microsomes |
a denotes mean ± standard deviation
2.2.2 Clinical Data¶
A literature search was performed to collect available clinical data on fluvoxamine in healthy adults.
The fluvoxamine PBPK model was developed using 26 different clinical studies with pharmacokinetic (PK) blood sampling. These studies include 1 study of 30 mg fluvoxamine administered intravenously (iv) as a single-dose, and 25 studies of fluvoxamine administered orally (po) in single- or multiple-doses. In the single-dose po studies fluvoxamine was administered in doses of 25 - 200 mg. In the multiple-dose po studies fluvoxamine was administered once (q.d.) or twice daily (b.i.d.), in doses of 10 - 150 mg per administration.
2.2.2.1 Model Building¶
The following studies were used for model building (training data):
| Publication | Arm / Treatment / Information used for model building |
|---|---|
| Japanese Society 2015 | Healthy Japanese adults with 30 mg as 60 min infusion or oral administration of 200 mg |
| de Vries 1993 | Healthy adults with oral administration of 25-100 mg |
| Orlando 2010 | Healthy adults with oral administration of 50 mg |
| Labellarte 2004 | Healthy CYP2D6 EM with oral administration of 50 mg twice a day |
| Spigset 1998 | Healthy CYP2D6 EM (80%) and PM (20%) with oral administration of doses between 12.5-100 mg twice a day |
| Fleishaker 1994 | Healthy adults with oral administration of 50 mg or 100 mg once daily |
2.2.2.2 Model Verification¶
The following studies were used for model verification:
| Publication | Arm / Treatment / Information used for model building |
|---|---|
| Christensen 2002 | Healthy CYP2D6 EM with oral administration of 10 mg or 25 mg twice a day and healthy CYP2D6 PM with oral administration of 10 mg or 25 mg once daily |
| Fukasawa 2006 | Healthy Japanese adults with single oral doses of 50 mg |
| Japanese Society 2015 | Healthy Japanese adults with single oral doses of 25-100 mg |
| Kunii 2005 | Healthy CYP2D6 EM with single oral doses of 50 mg |
| Spigset 1995 | Healthy smokers or non-smokers with oral administration of 50 mg as single dose |
| Spigset 1997 | Healthy CYP2D6 EM or PM with oral administration of 50 mg as single dose |
| van Harten 1991 | Healthy adults with oral administration of 50 mg as single dose |
| de Vries 1992 | Healthy adults with oral administration of 50 mg twice a day |
| Bahrami 2007 | Healthy adults with oral administration of 100 mg as single dose |
| de Bree 1983 | Healthy adults with oral administration of 100 mg as single dose |
2.3 Model Parameters and Assumptions¶
2.3.1 Absorption¶
Since a rapid dissolution and absorption was assumed for tablet as well as capsule formulation, the drug formulation was implemented as solution.
The specific intestinal permeability was identified during parameter identification.
2.3.2 Distribution¶
It is described in literature that fluvoxamine is moderately bound to plasma proteins (77%, Claassen 1983). This value was implemented in PK-Sim®. The protein binding partner was set to unknown.
An important parameter influencing the distribution of a compound is lipophilicity. To accurately describe the distribution of fluvoxamine, logP was optimized during parameter identification to match observed clinical data.
After testing the available organ-plasma partition coefficient and cell permeability calculation methods built in PK-Sim, observed clinical data was best described by choosing the partition coefficient calculation by Schmitt and cellular permeability calculation by PK-Sim Standard.
2.3.3 Metabolism and Elimination¶
The final model applies metabolism by CYP1A2, CYP2D6 and glomerular filtration. The metabolic processes by CYP1A2 and CYP2D6 were described by Michaelis-Menten kinetics. The Michaelis-Menten constant Km for CYP2D6 metabolism was fixed according to literature values, other parameters were identified during parameter identification.
To distinguish between fluvoxamine metabolism in CYP2D6 extensive metabolizers (EM) and poor metabolizers (PM), the CYP2D6 catalytic rate constant kcat of PMs was set to zero. This assumption was made because CYP2D6 PMs were characterized by absent CYP2D6 enzymatic activity Crews 2014, which results in a predicted 1.5-fold increase of the fluvoxamine AUC in CYP2D6 PMs compared with CYP2D6 EMs.
Smoking is the strongest known inducer of CYP1A2 and results in higher metabolism of CYP1A2 substrates Zhou 2009. As no detailed information on the frequency, duration, and amount of smoking was available from literature, the induction of CYP1A2 was implemented as a static 1.38-fold increase in enzyme activity. This factor was optimized based on the study of Spigset et al. (Spigset 1995) resulting in a 39% reduction of the fluvoxamine AUC in smokers.
2.3.4 Enzyme inhibition¶
To describe the inhibition of CYP1A2 by fluvoxamine, the reported Ki value of 11 nmol/L Karjalainen 2008 was corrected for fluvoxamine binding in the in vitro test system as recommended by Yao 2001 and a value of 10 nmol/L was then used for both Ki_c and Ki_u to describe mixed-type inhibition in the PBPK model.
To describe the inhibition of CYP3A4 by fluvoxamine, the reported Ki value of 1.6 µmol/L (Olesen 2000) was included in the model.
2.3.5 Automated Parameter Identification¶
This is the result of the final parameter identification.
| Model Parameter | Optimized Value | Unit |
|---|---|---|
logP |
3.57 | log units |
Km (CYP1A2) |
7.35 | nmol/L |
kcat (CYP1A2) non-smokers |
0.016 | 1/min |
kcat (CYP1A2) smokers |
0.022 | 1/min |
kcat (CYP2D6) extensive metabolizers (EM) |
110.56 | 1/min |
Specific intestinal permeability |
2.74 E-6 | dm/min |
3 Results and Discussion¶
The PBPK model for fluvoxamine was developed and verified with clinical pharmacokinetic data.
The model was evaluated covering data from studies including in particular
- intravenous and oral administration
- single and multiple doses
- a dose range from 25 mg to 200 mg
- subjects phenotyped as CYP2D6 extensive metabolizers (EM) and poor metabolizers (PM)
- smokers and non-smokers
The model quantifies metabolism via CYP1A2 and CYP2D6 and the effect of smoking and different CYP2D6 phenotypes on fluvoxamine metabolism.
The next sections show:
- the final model parameters for the building blocks: Section 3.1.
- the overall goodness of fit: Section 3.2.
- simulated vs. observed concentration-time profiles for the clinical studies used for model building and for model verification: Section 3.3.
3.1 Final input parameters¶
The compound parameter values of the final PBPK model are illustrated below.
Compound: Fluvoxamine¶
Parameters¶
| Name | Value | Value Origin | Alternative | Default |
|---|---|---|---|---|
| Solubility at reference pH | 14.66 mg/ml | Measurement | True | |
| Reference pH | 7 | Measurement | True | |
| Lipophilicity | 3.5726507829 Log Units | Parameter Identification | Measurement | True |
| Fraction unbound (plasma, reference value) | 0.23 | Publication-Claassen et al., Review of the animal pharmacology and pharmacokinetics of fluvoxamine. Br. J. Clin. Pharmacol. 15, 349S-355S (1983). | Measurement | True |
| Specific intestinal permeability (transcellular) | 2.7380788903E-06 dm/min | Parameter Identification | Fitted | True |
| F | 3 | |||
| Is small molecule | Yes | |||
| Molecular weight | 318.335 g/mol | |||
| Plasma protein binding partner | Unknown |
Calculation methods¶
| Name | Value |
|---|---|
| Partition coefficients | Schmitt |
| Cellular permeabilities | PK-Sim Standard |
Processes¶
Metabolizing Enzyme: CYP1A2-Fit¶
Molecule: CYP1A2
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 0 pmol/min/mg mic. protein | |
| Content of CYP proteins in liver microsomes | 45 pmol/mg mic. protein | Unknown |
| Km | 0.0073460807948 µmol/l | |
| kcat | 0.0155447966 1/min | Unknown |
Metabolizing Enzyme: CYP2D6-Miura2007¶
Molecule: CYP2D6
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax/recombinant enzyme | 0.69 pmol/min/pmol rec. enzyme | |
| Km | 76.3 µmol/l | |
| kcat | 110.5561921693 1/min | Parameter Identification |
Systemic Process: Glomerular Filtration-4% Urine¶
Species: Human
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| GFR fraction | 1 |
Inhibition: CYP1A2-Karjalainen2008/Yao2001¶
Molecule: CYP1A2
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Ki_c | 10 nmol/l | Publication-In Vitro-Karjalainen et al. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin. Pharmacol. Toxicol. 103, 157–65 (2008) and Yao, C. et al. Fluvoxamine-theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin. Pharmacol. Ther. 70, 415–24 (2001) |
| Ki_u | 10 nmol/l | Publication-In Vitro-Karjalainen et al. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin. Pharmacol. Toxicol. 103, 157–65 (2008) and Yao, C. et al. Fluvoxamine-theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin. Pharmacol. Ther. 70, 415–24 (2001) |
Inhibition: CYP3A4-Olesen2000/Yao2001¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Ki | 1.6 µmol/l | Publication-In Vitro-Olesen et al. Fluvoxamine-Clozapine drug interaction: inhibition in vitro of five cytochrome P450 isoforms involved in clozapine metabolism. J. Clin. Psychopharmacol. 20, 35–42 (2000) and Yao, C. et al. Fluvoxamine-theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin. Pharmacol. Ther. 70, 415–24 (2001) |
Formulation: Solution¶
Type: Dissolved
3.2 Diagnostics Plots¶
Below you find the goodness-of-fit visual diagnostic plots for the PBPK model performance of all data used presented in Section 2.2.2.
The first plot shows observed versus simulated plasma concentration, the second weighted residuals versus time.
Table 3-1: GMFE for Goodness of fit plot for concentration in plasma
| Group | GMFE |
|---|---|
| test dataset | 1.62 |
| training dataset | 1.21 |
| All | 1.40 |
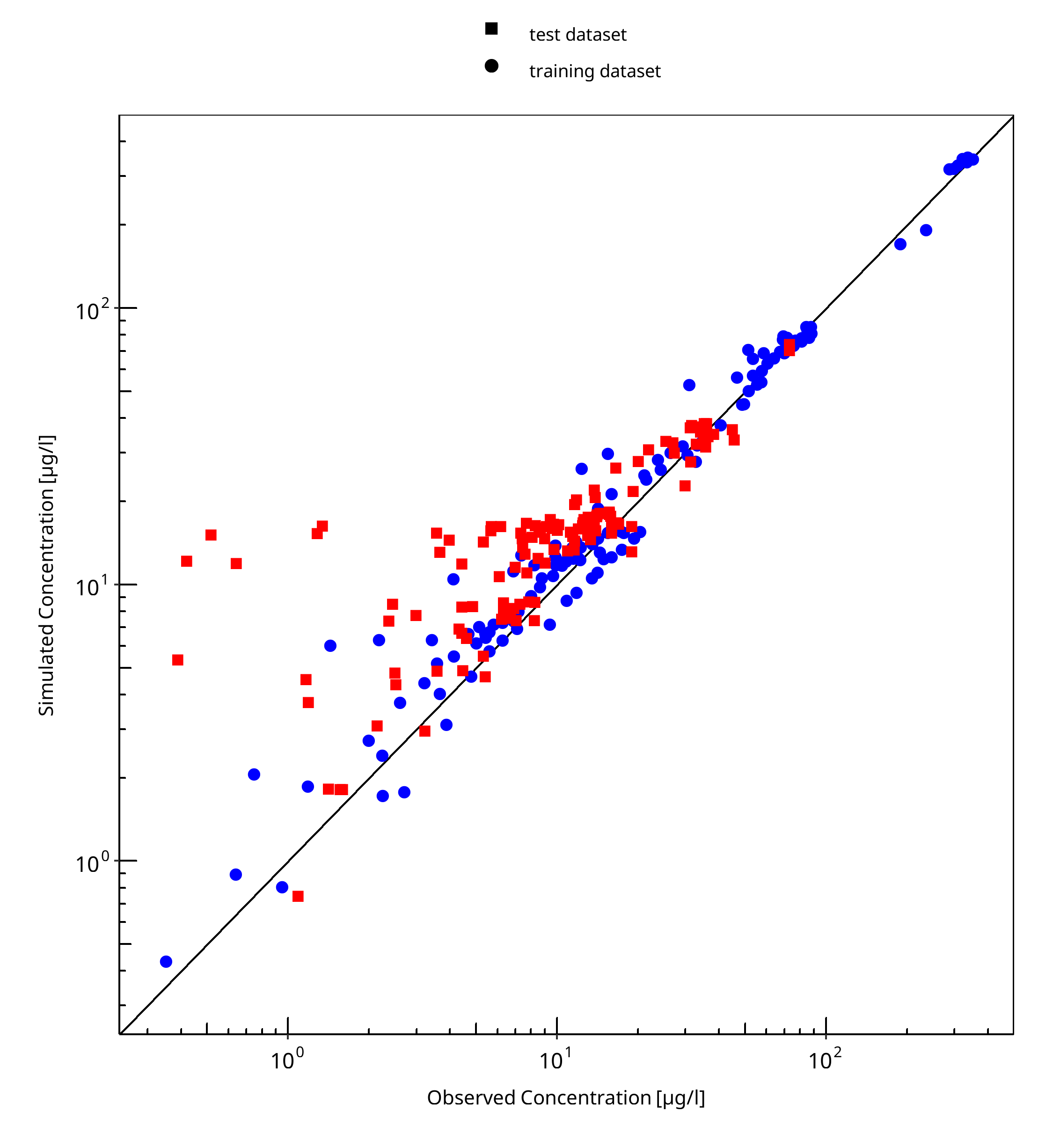
Figure 3-1: Goodness of fit plot for concentration in plasma
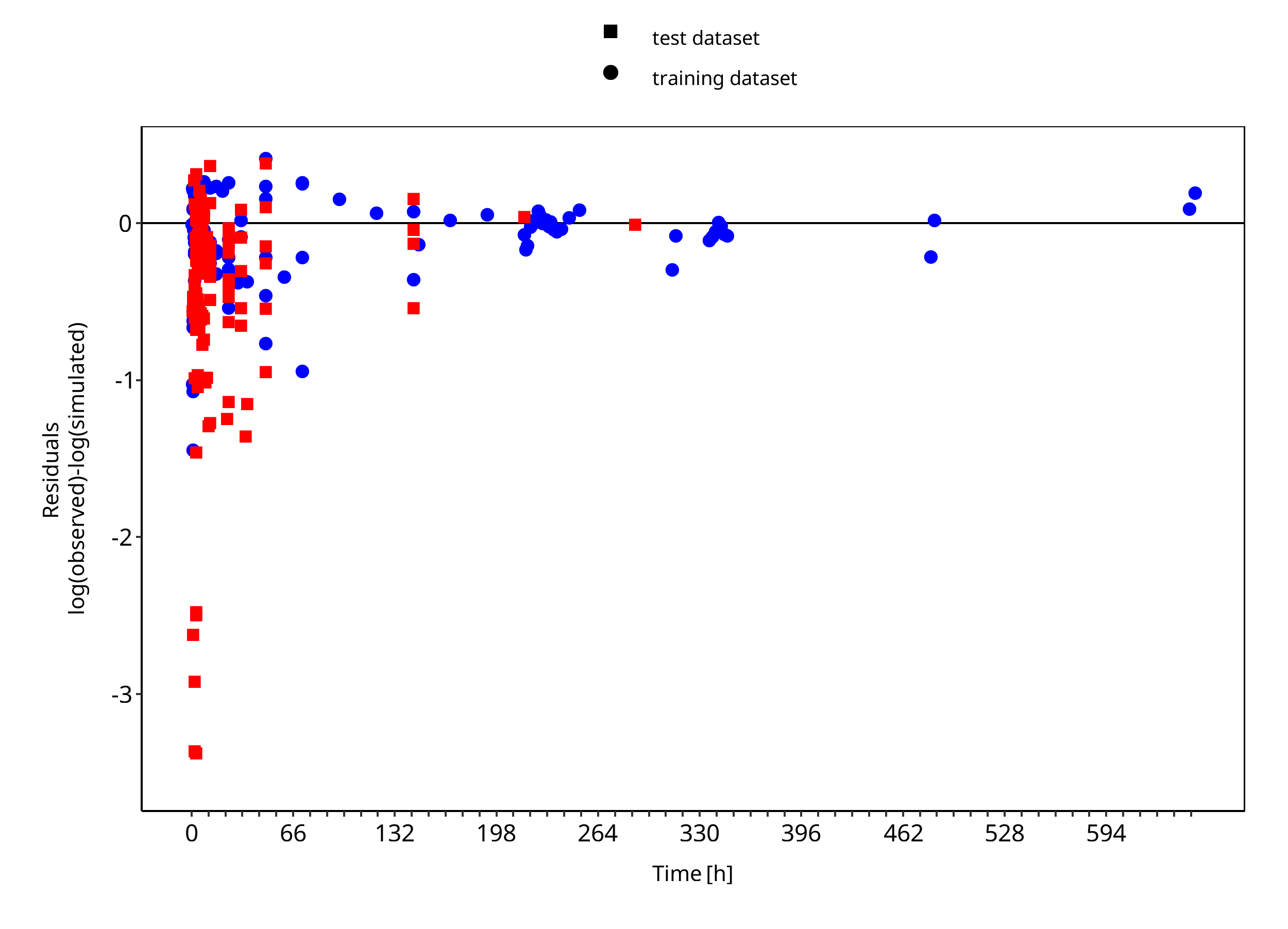
Figure 3-2: Goodness of fit plot for concentration in plasma
3.3 Concentration-Time Profiles¶
Simulated versus observed concentration-time profiles of all data listed in Section 2.2.2 are presented below.
3.3.1 Model Building¶
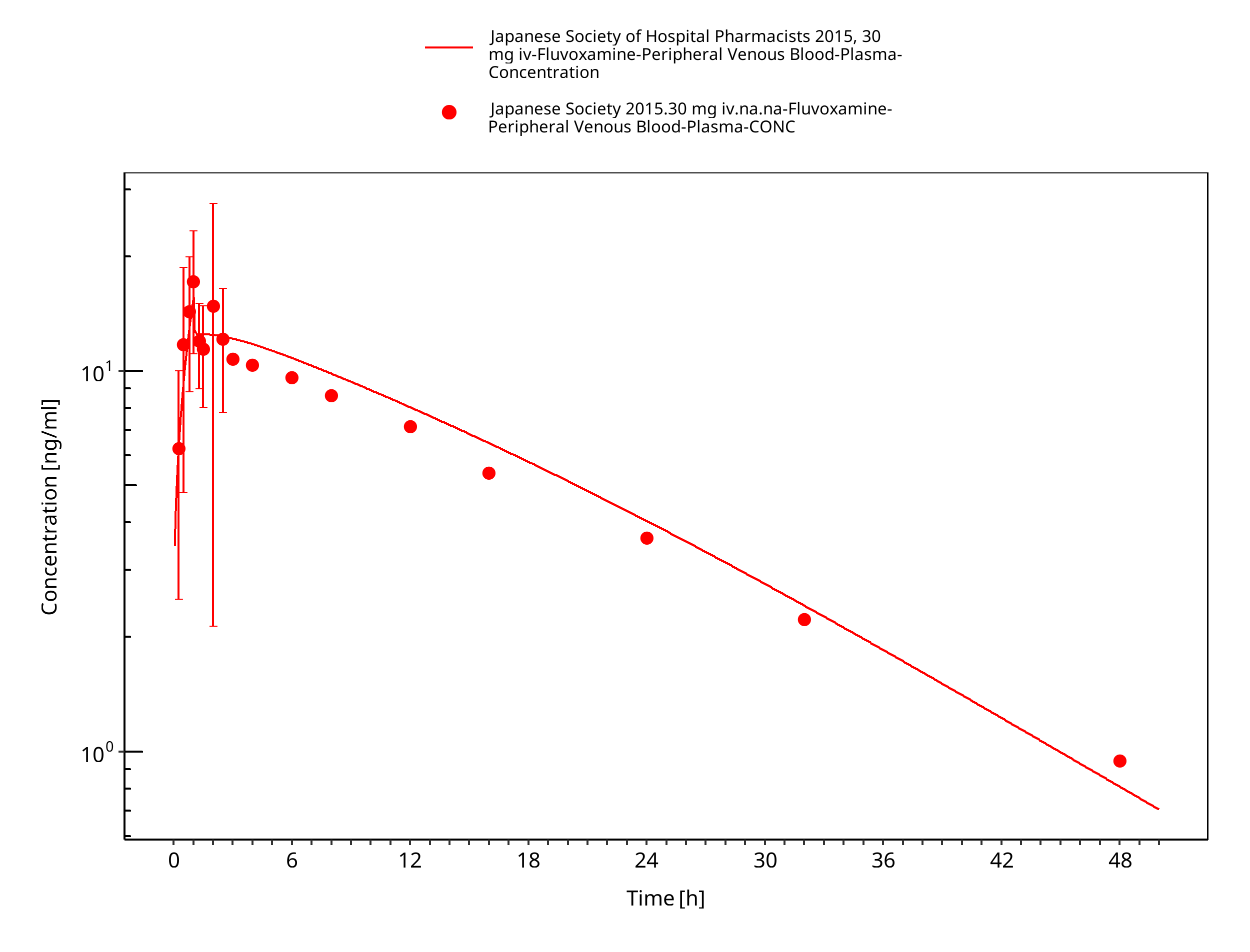
Figure 3-3: Time Profile Analysis
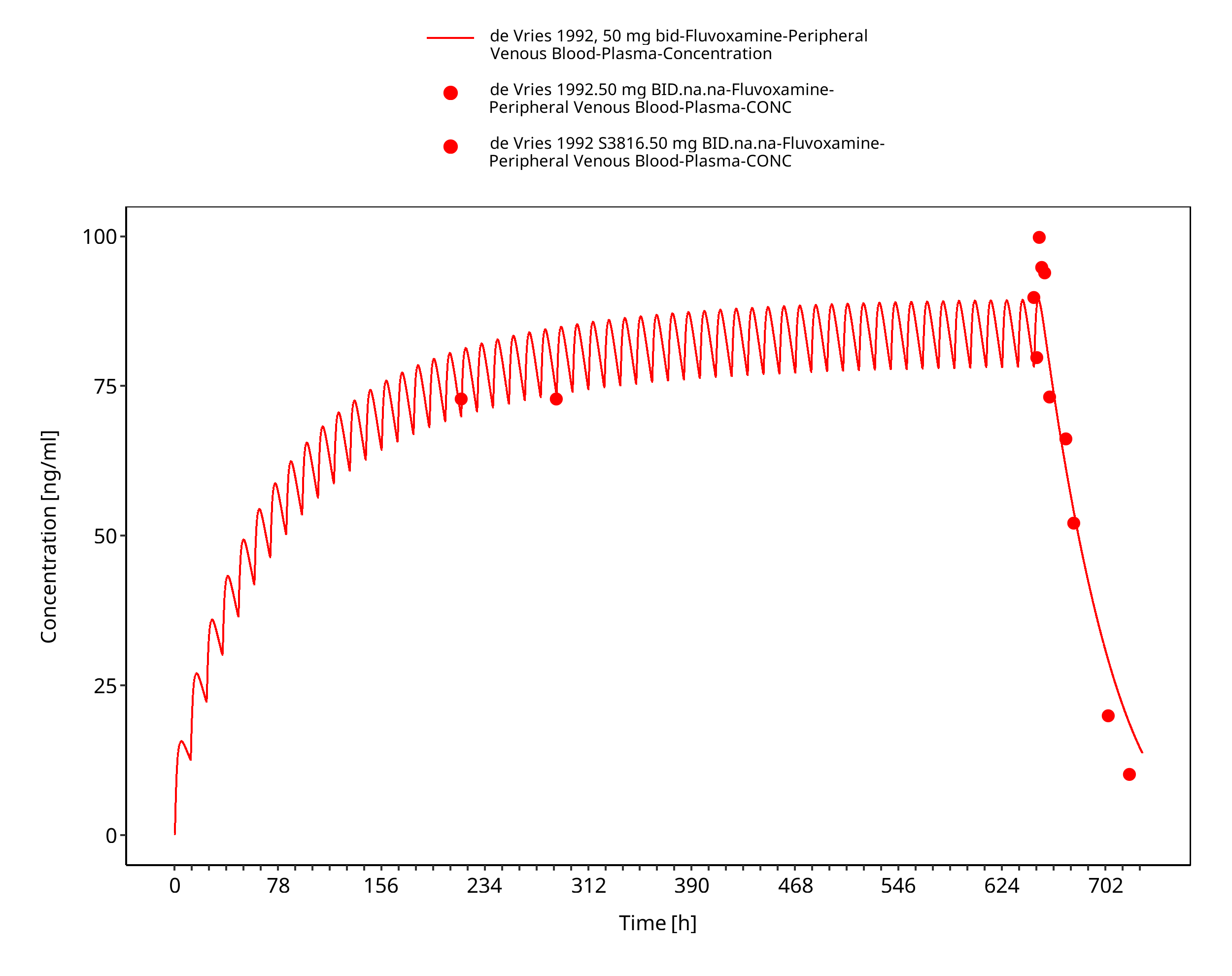
Figure 3-4: Time Profile Analysis
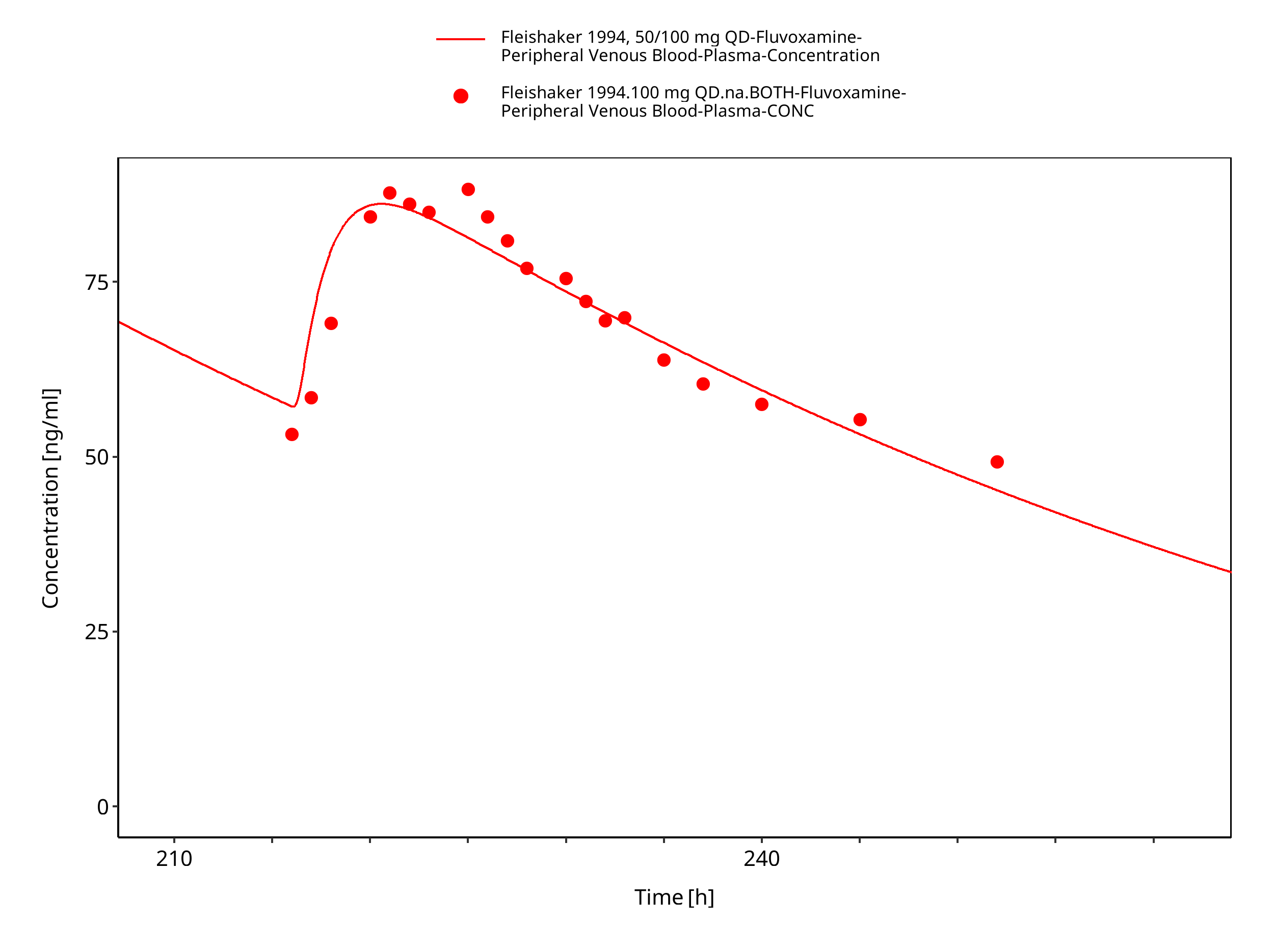
Figure 3-5: Time Profile Analysis
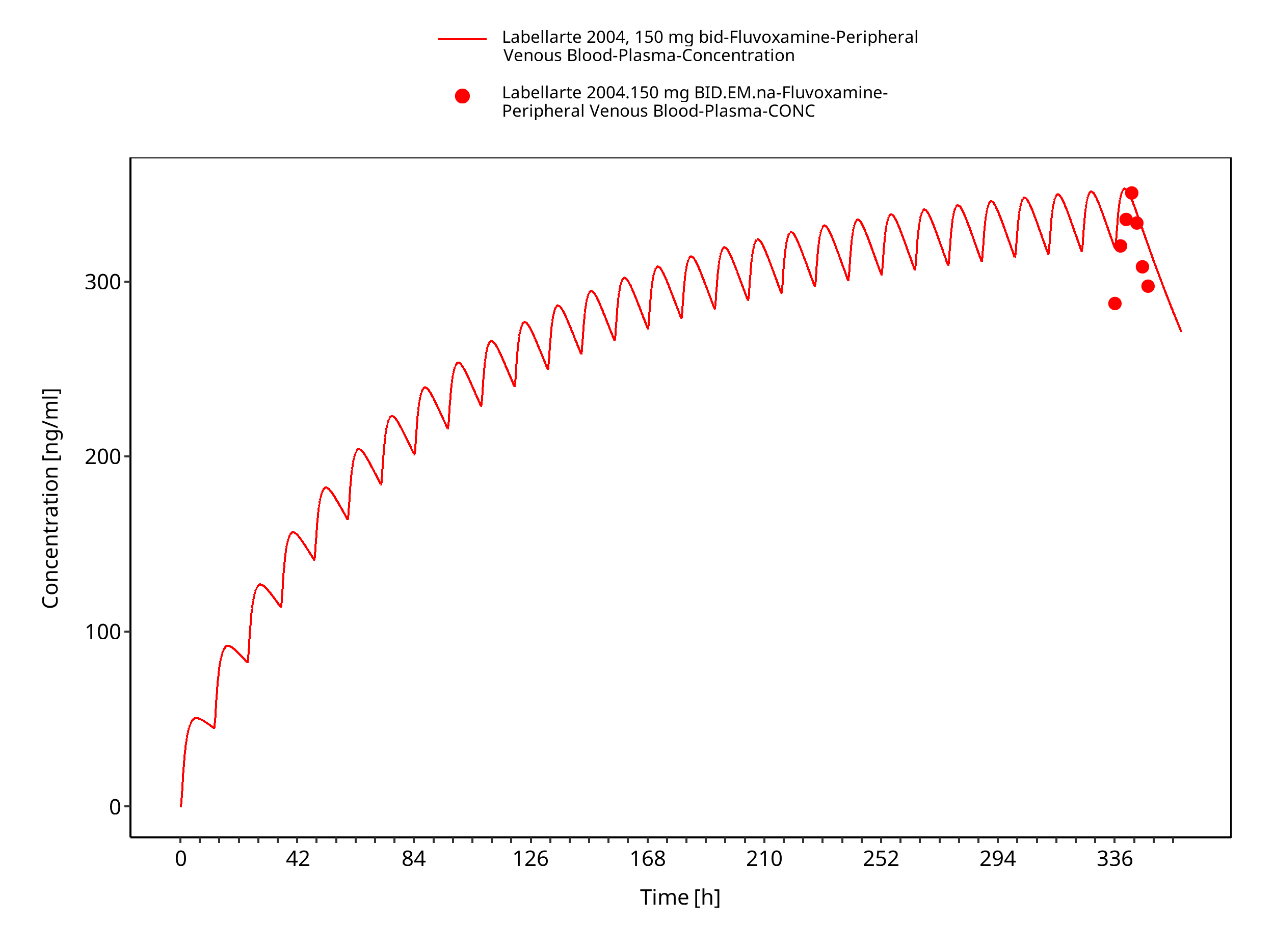
Figure 3-6: Time Profile Analysis
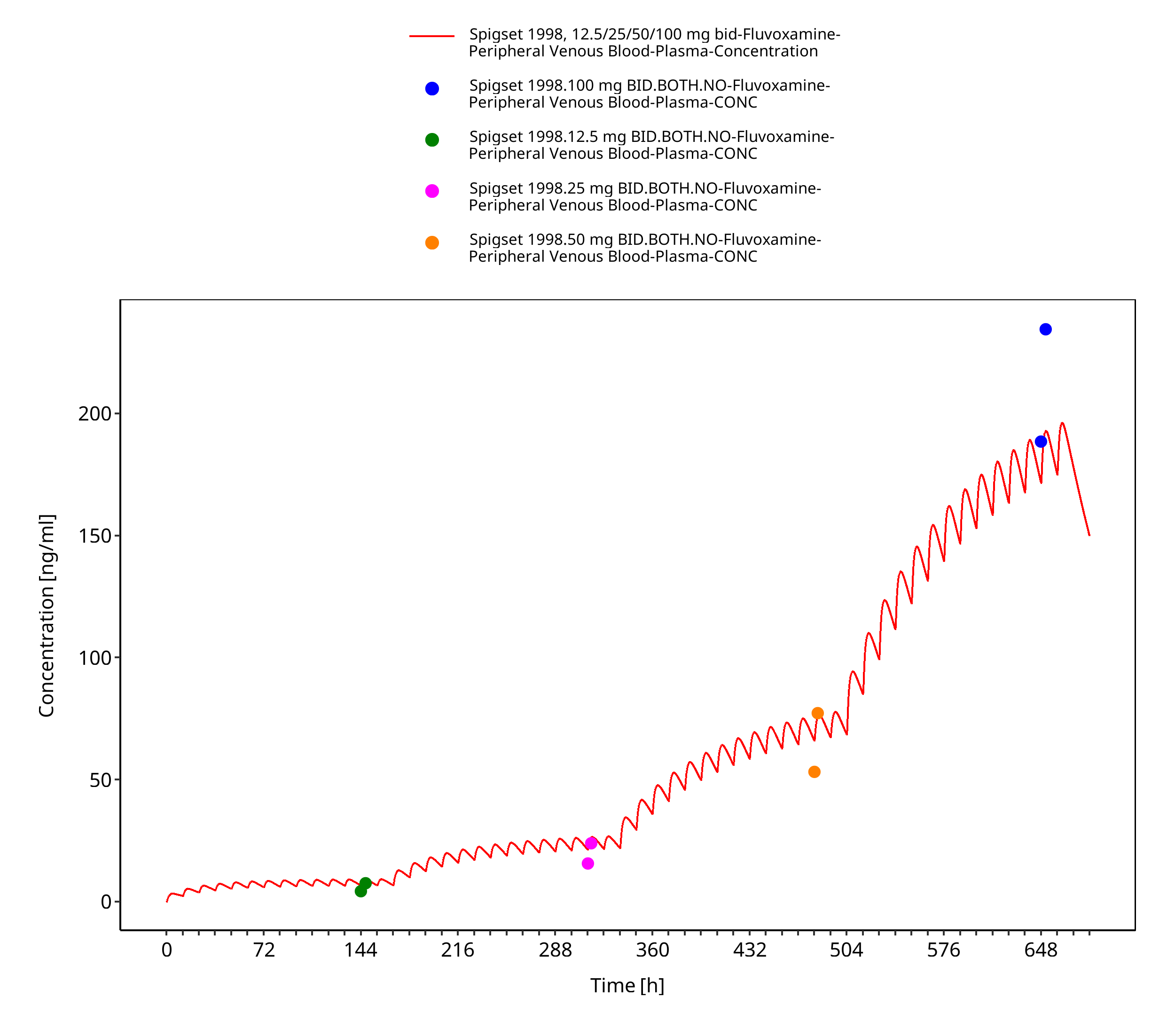
Figure 3-7: Time Profile Analysis
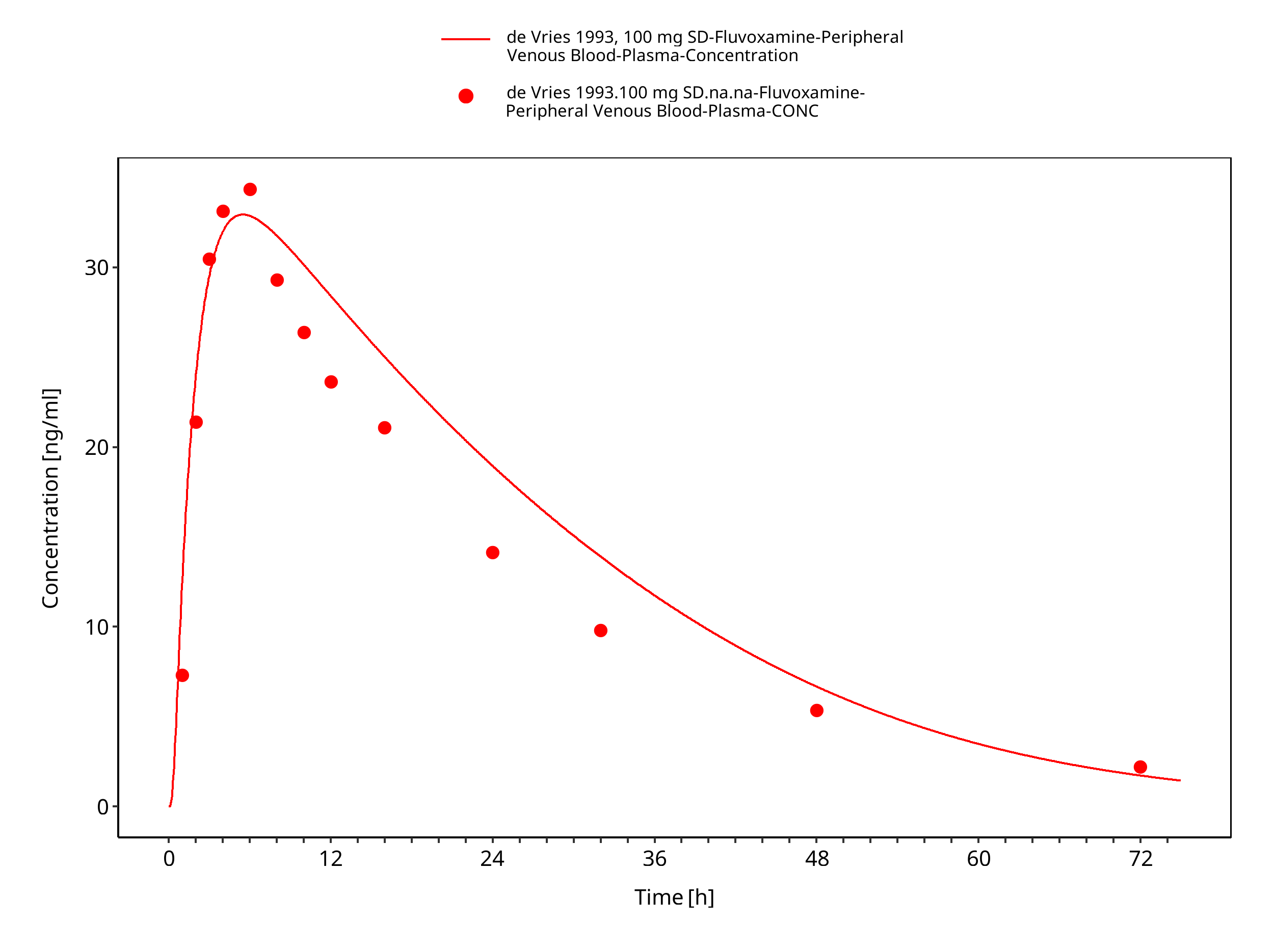
Figure 3-8: Time Profile Analysis
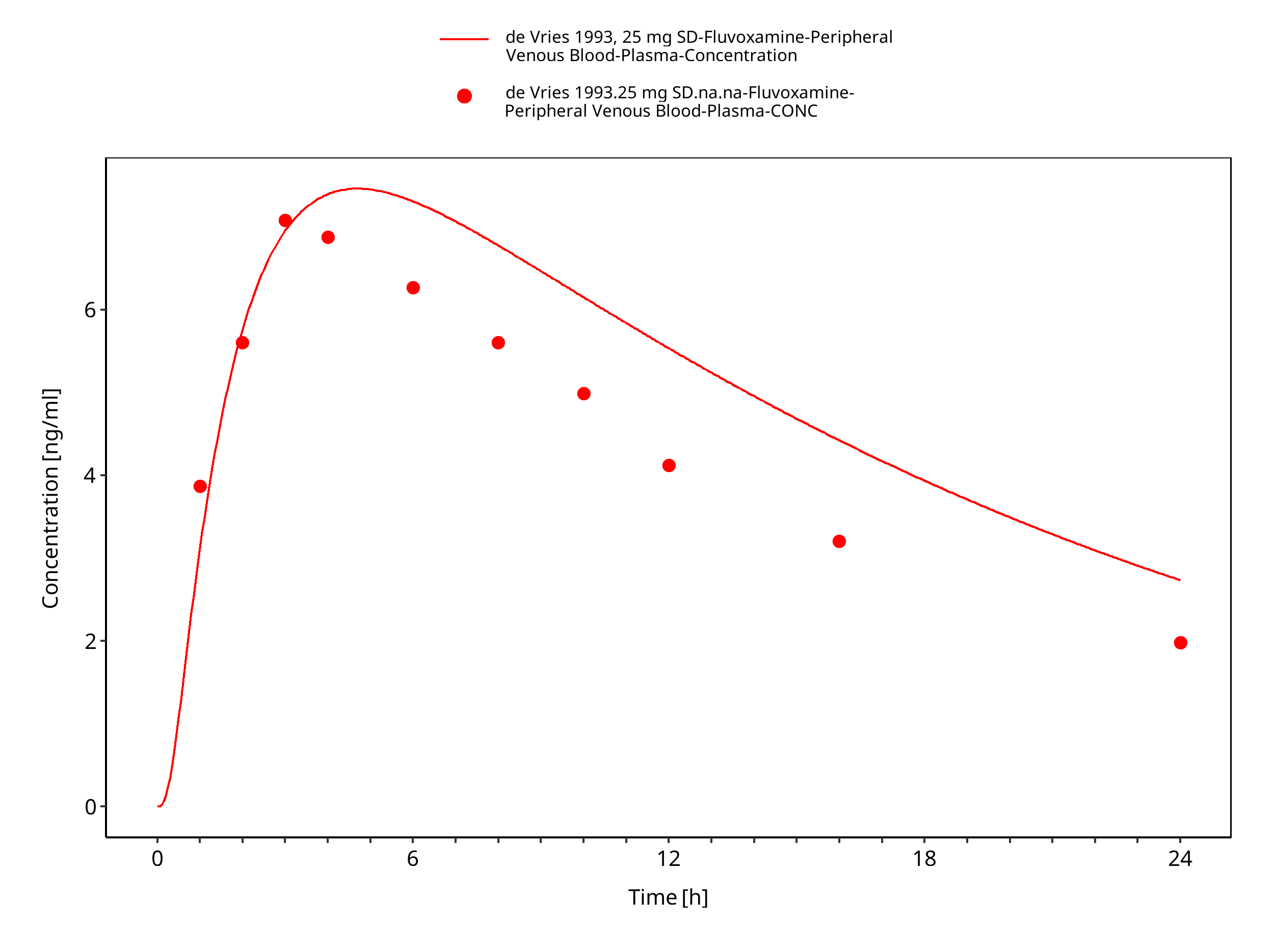
Figure 3-9: Time Profile Analysis
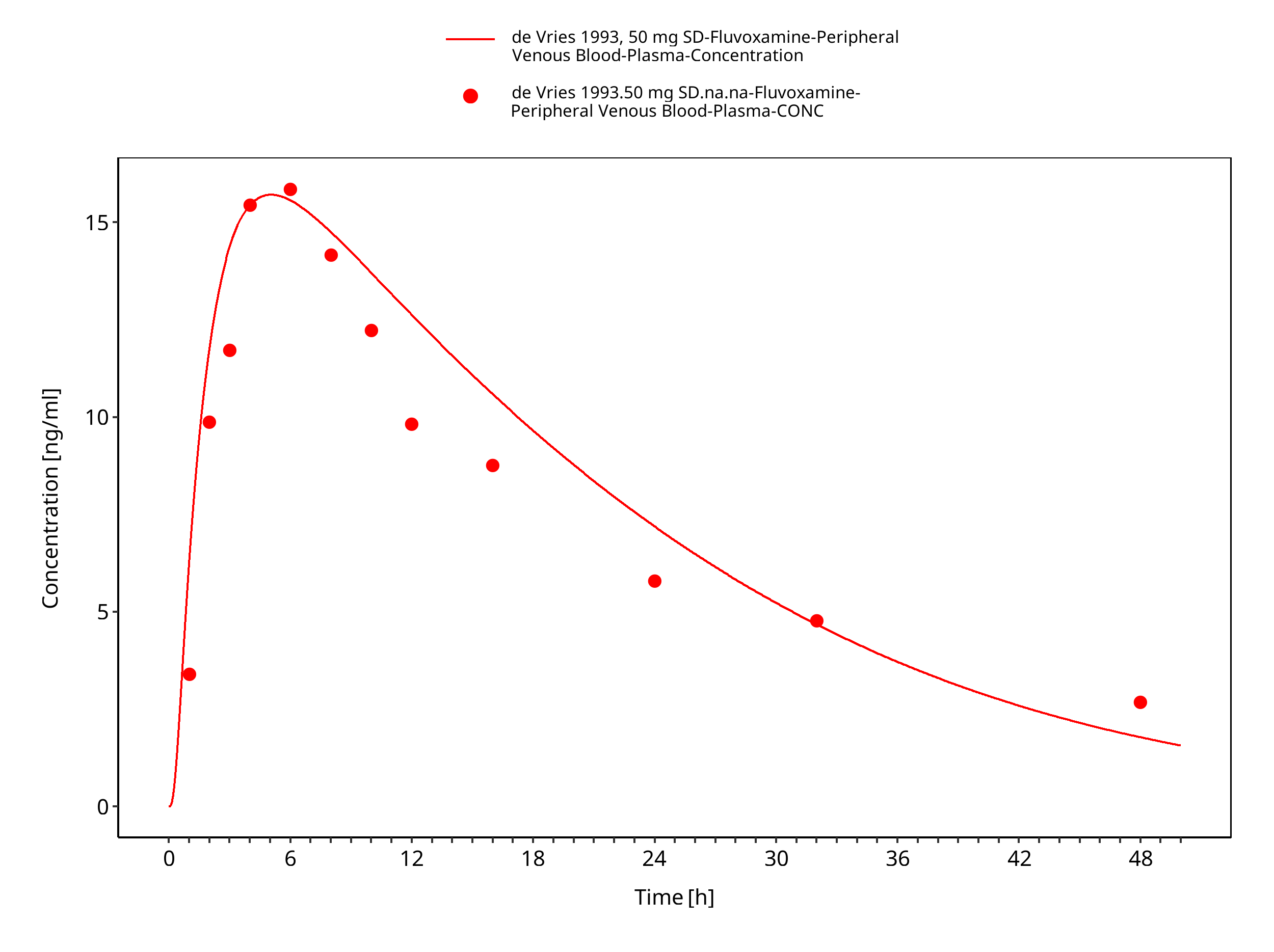
Figure 3-10: Time Profile Analysis
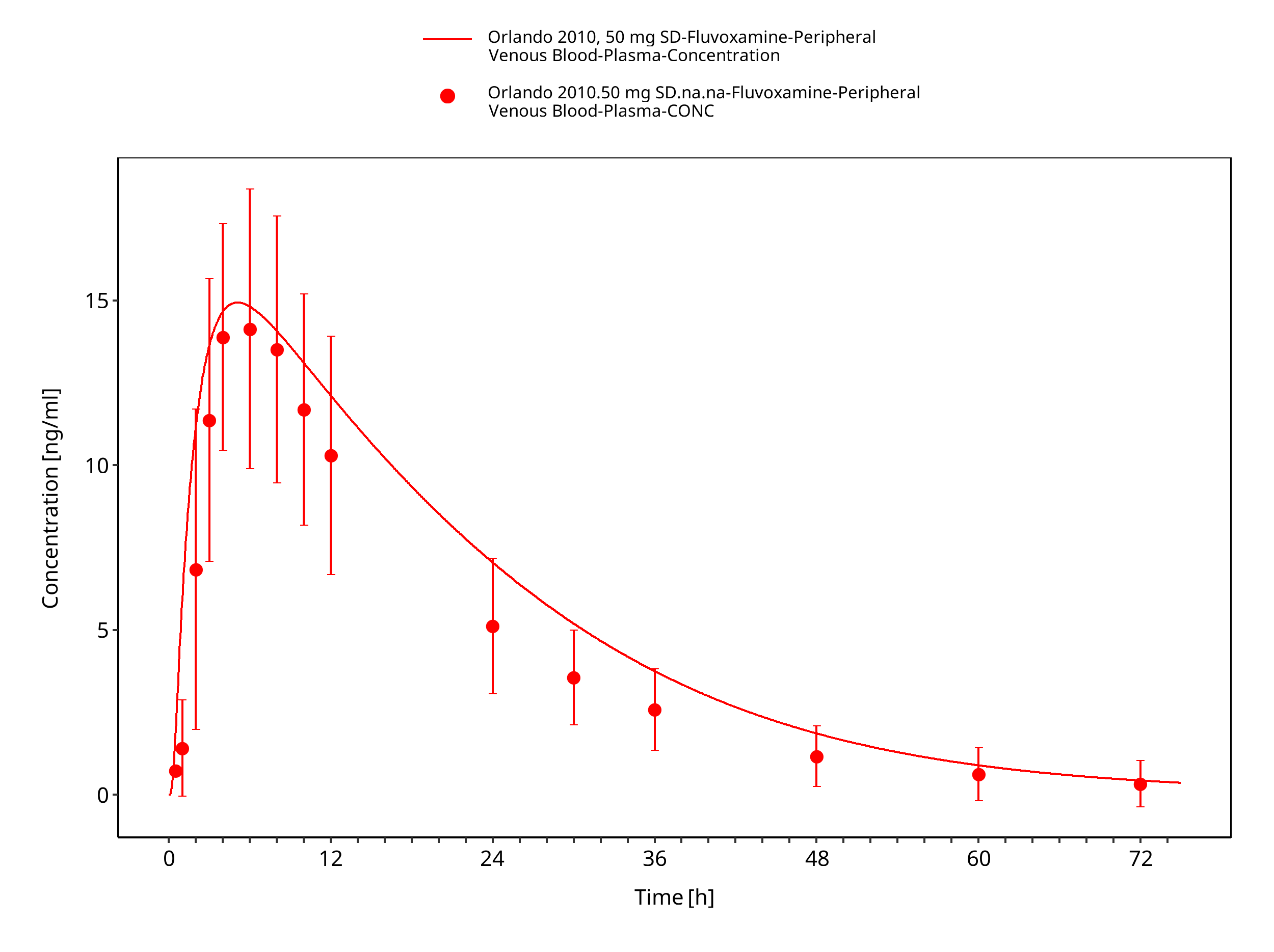
Figure 3-11: Time Profile Analysis
3.3.2 Model Verification¶
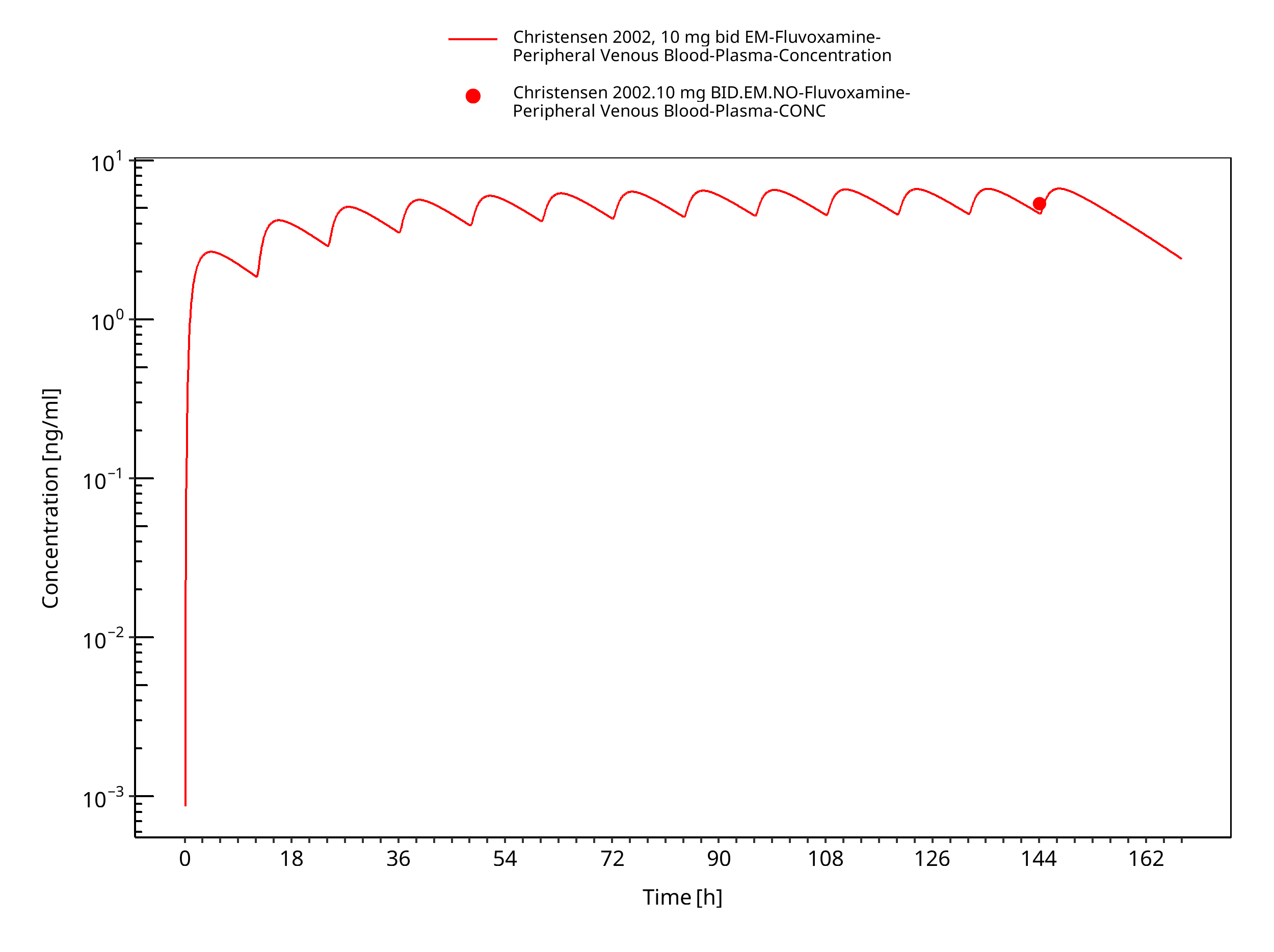
Figure 3-12: Time Profile Analysis
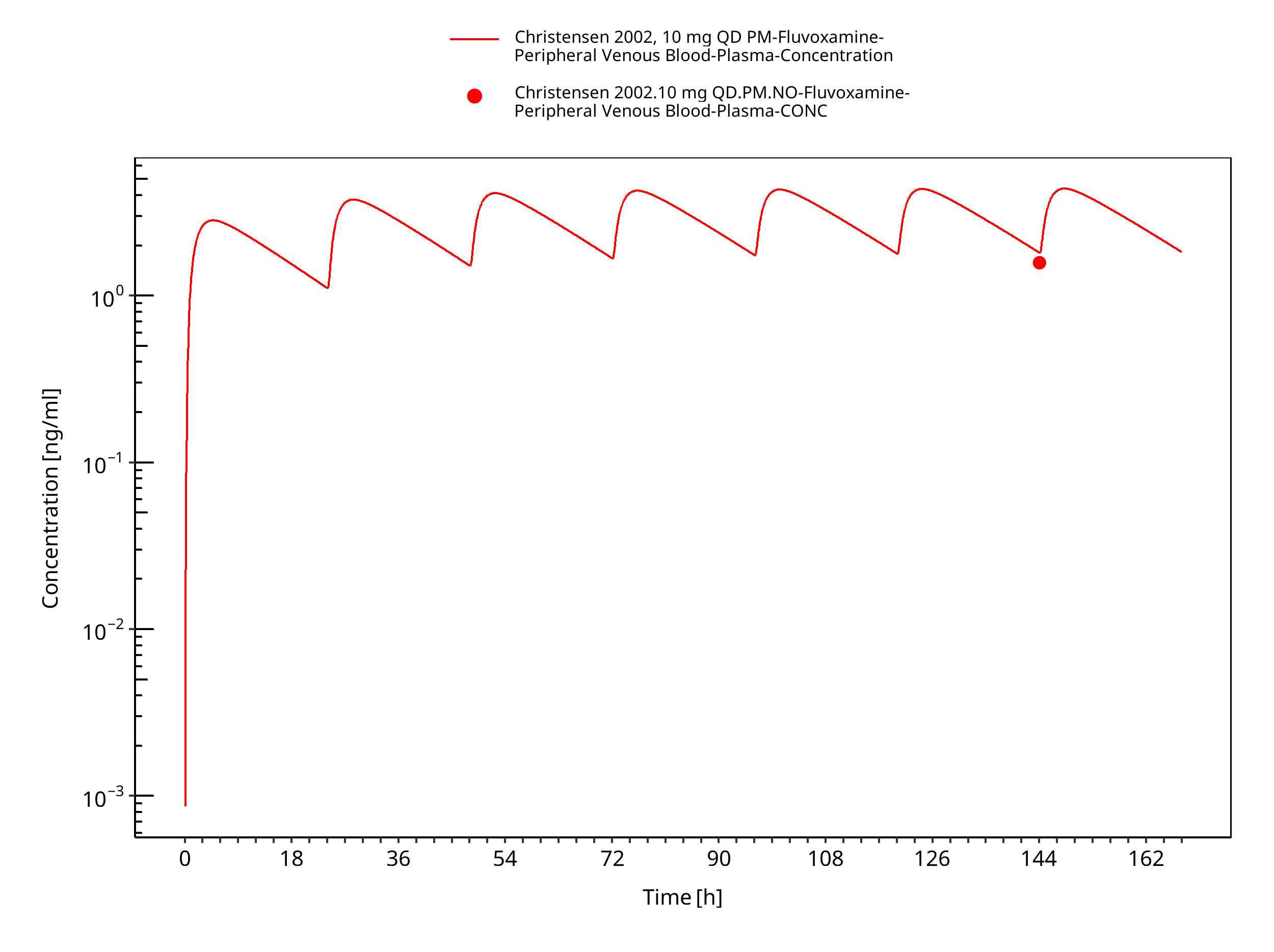
Figure 3-13: Time Profile Analysis
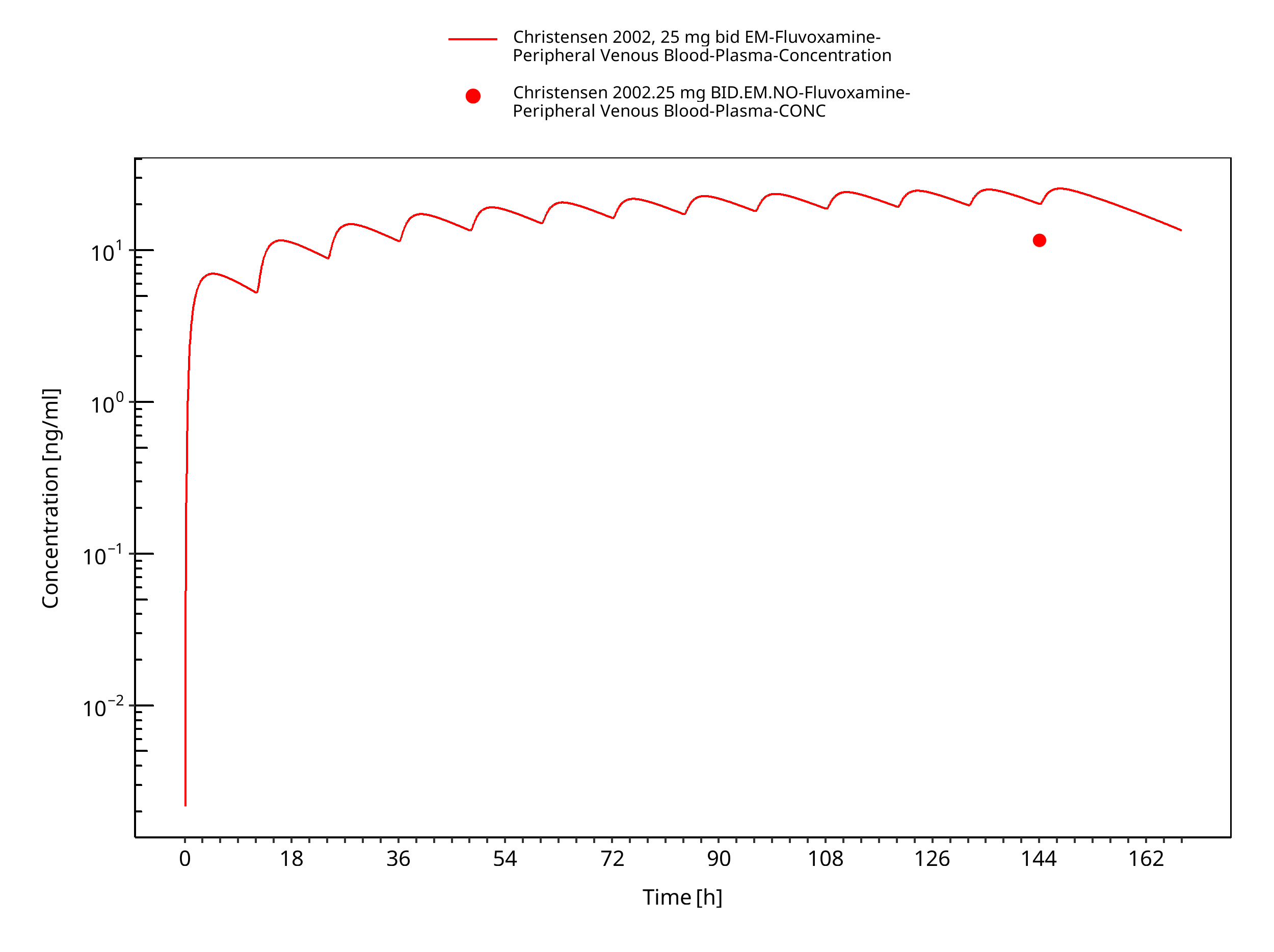
Figure 3-14: Time Profile Analysis
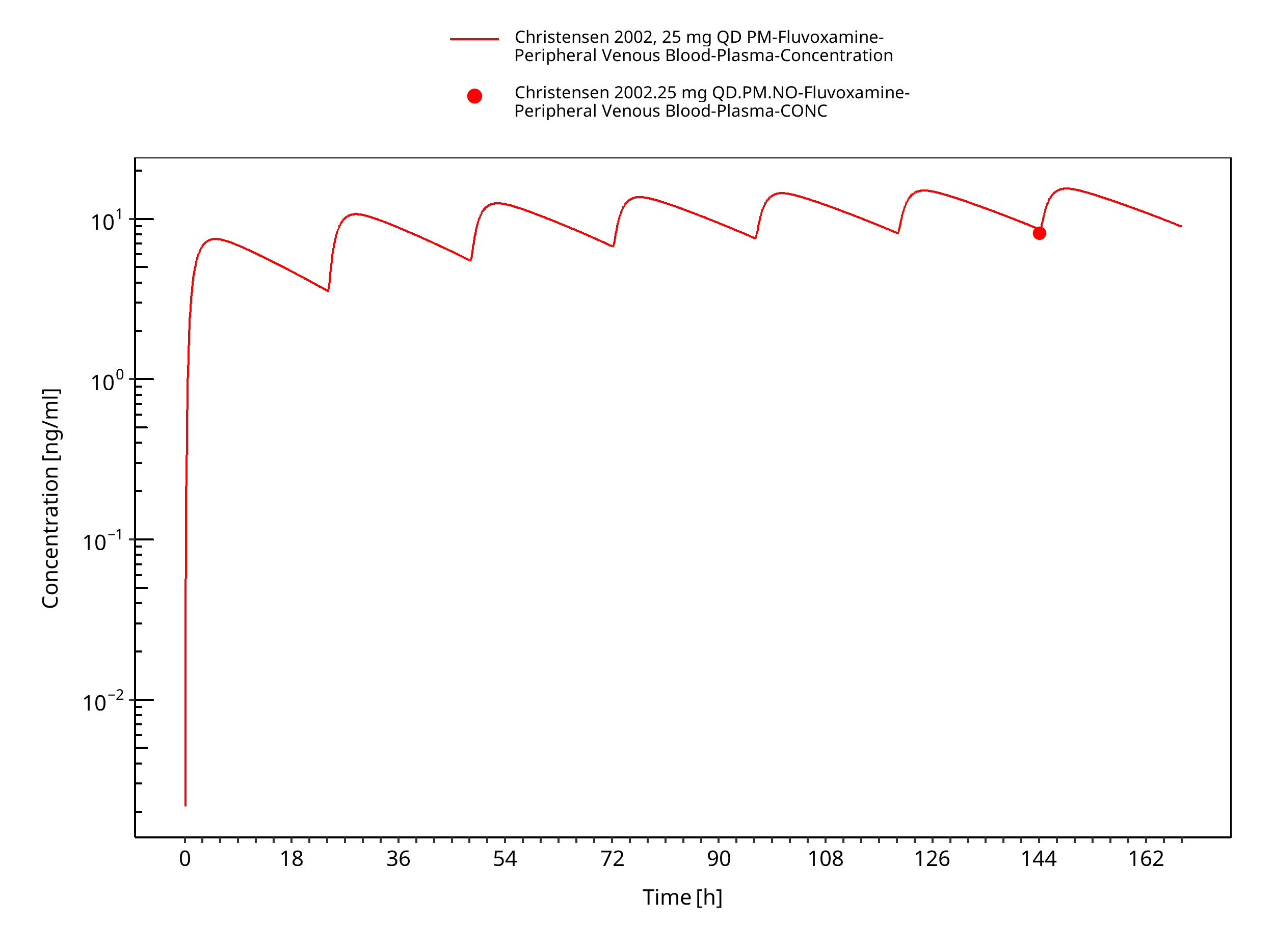
Figure 3-15: Time Profile Analysis
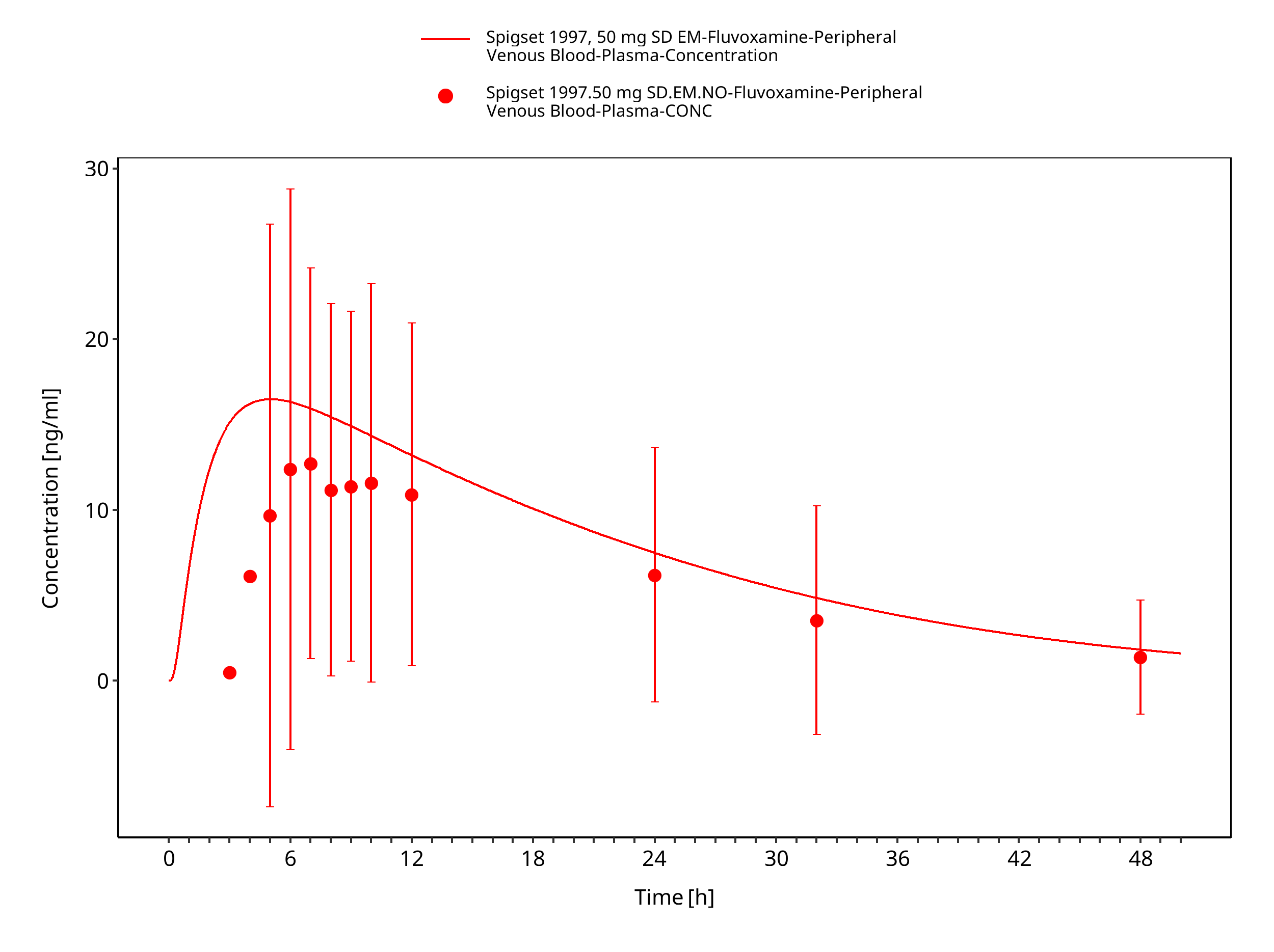
Figure 3-16: Time Profile Analysis
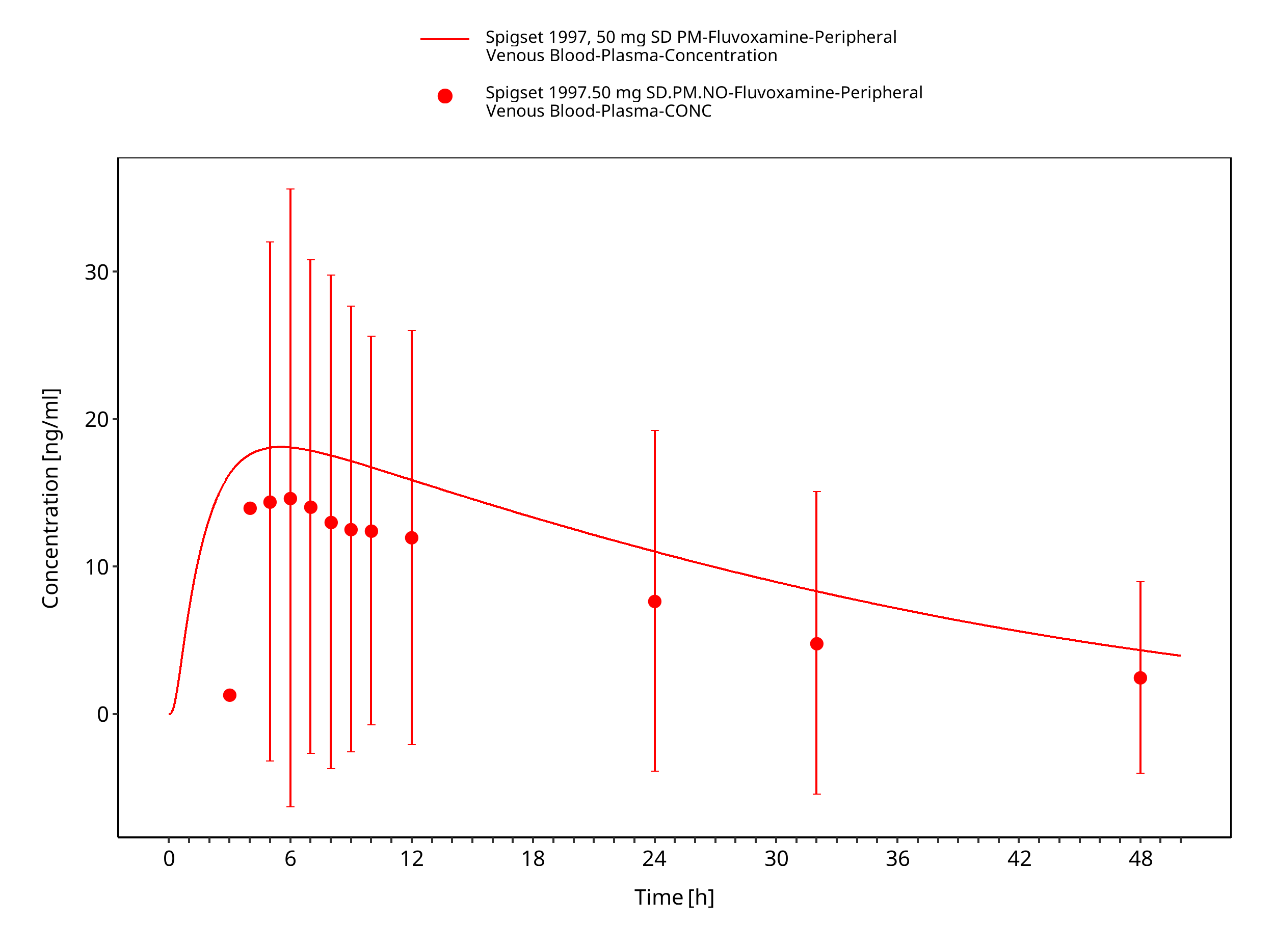
Figure 3-17: Time Profile Analysis
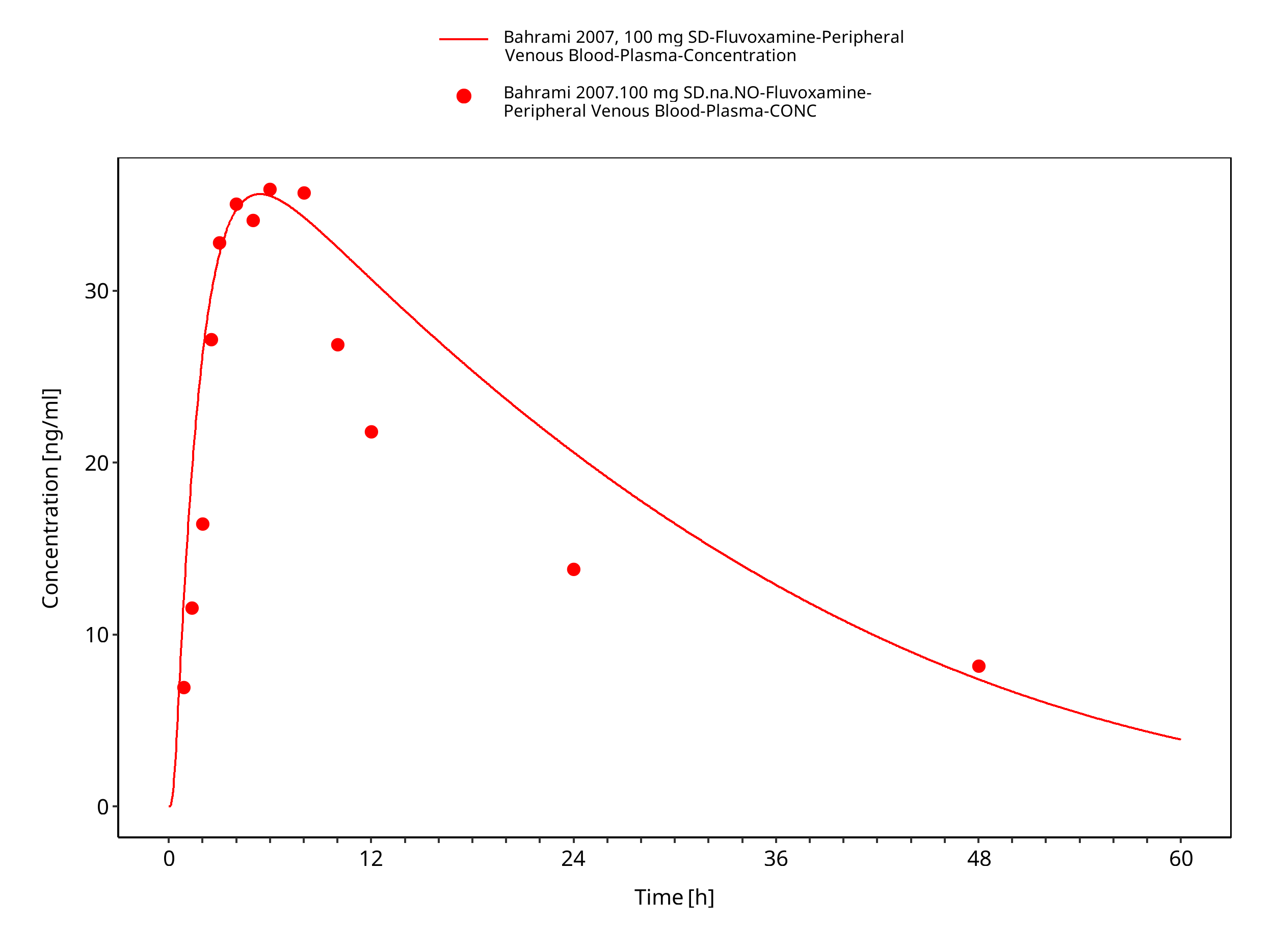
Figure 3-18: Time Profile Analysis
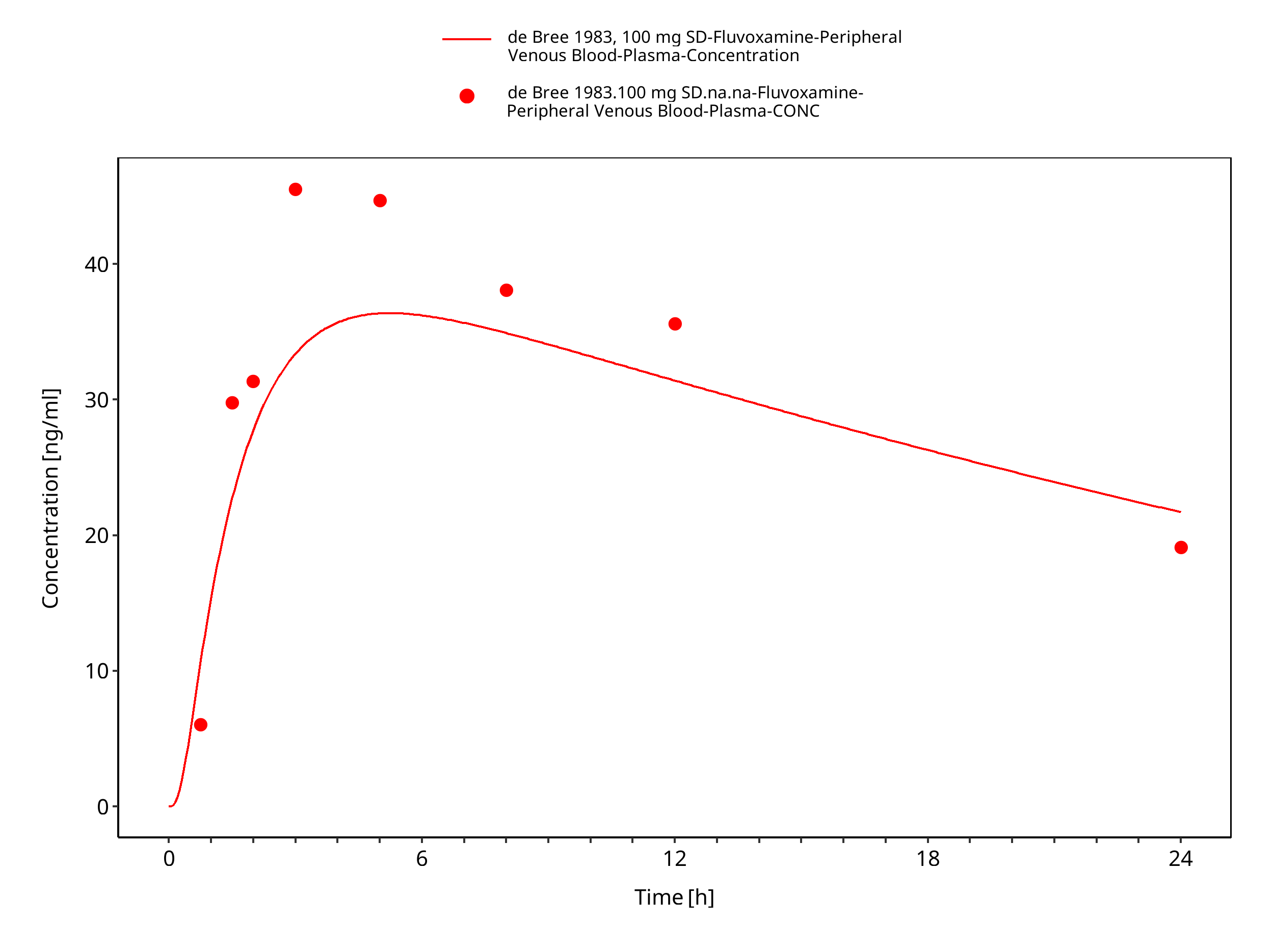
Figure 3-19: Time Profile Analysis
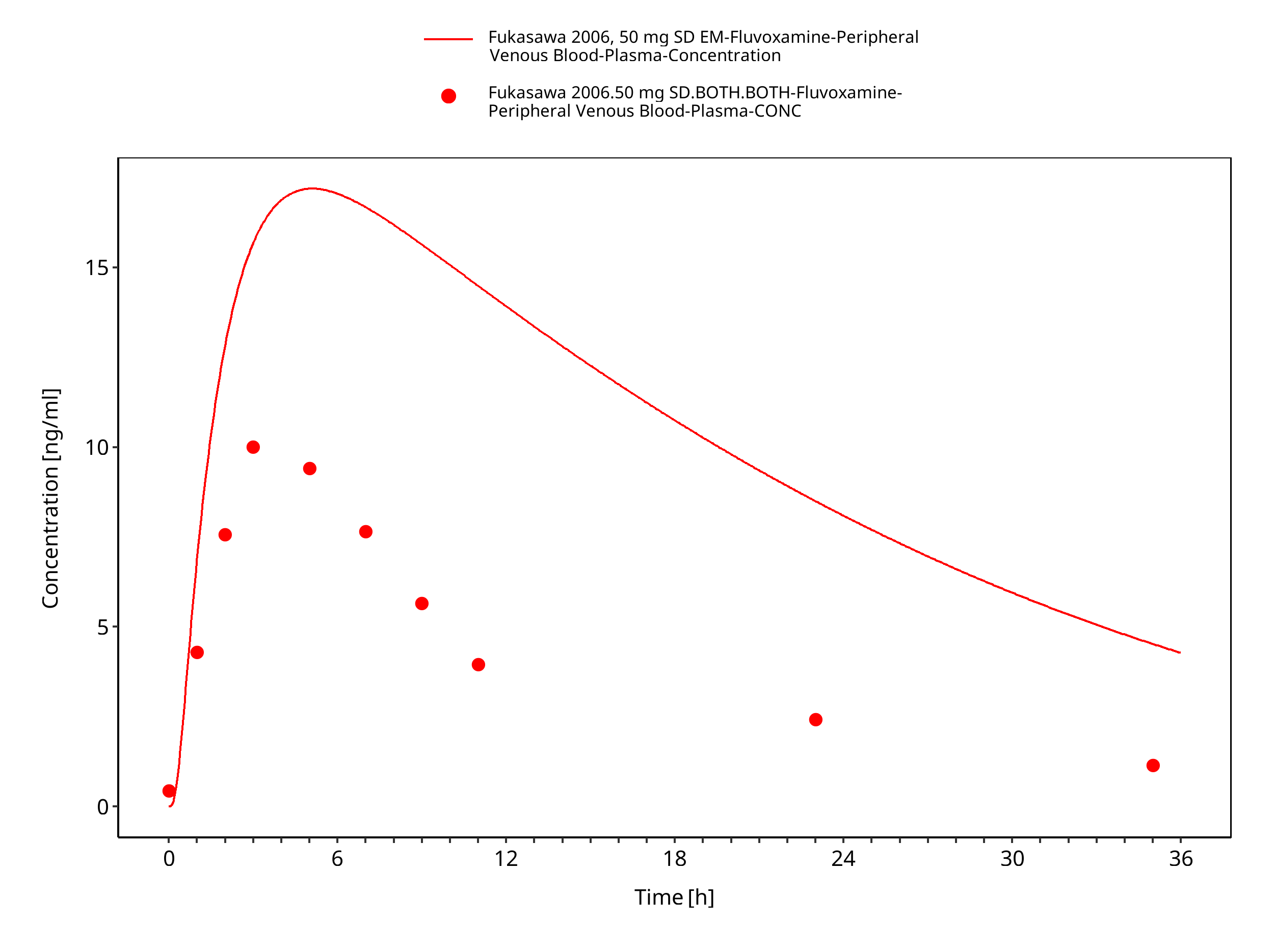
Figure 3-20: Time Profile Analysis
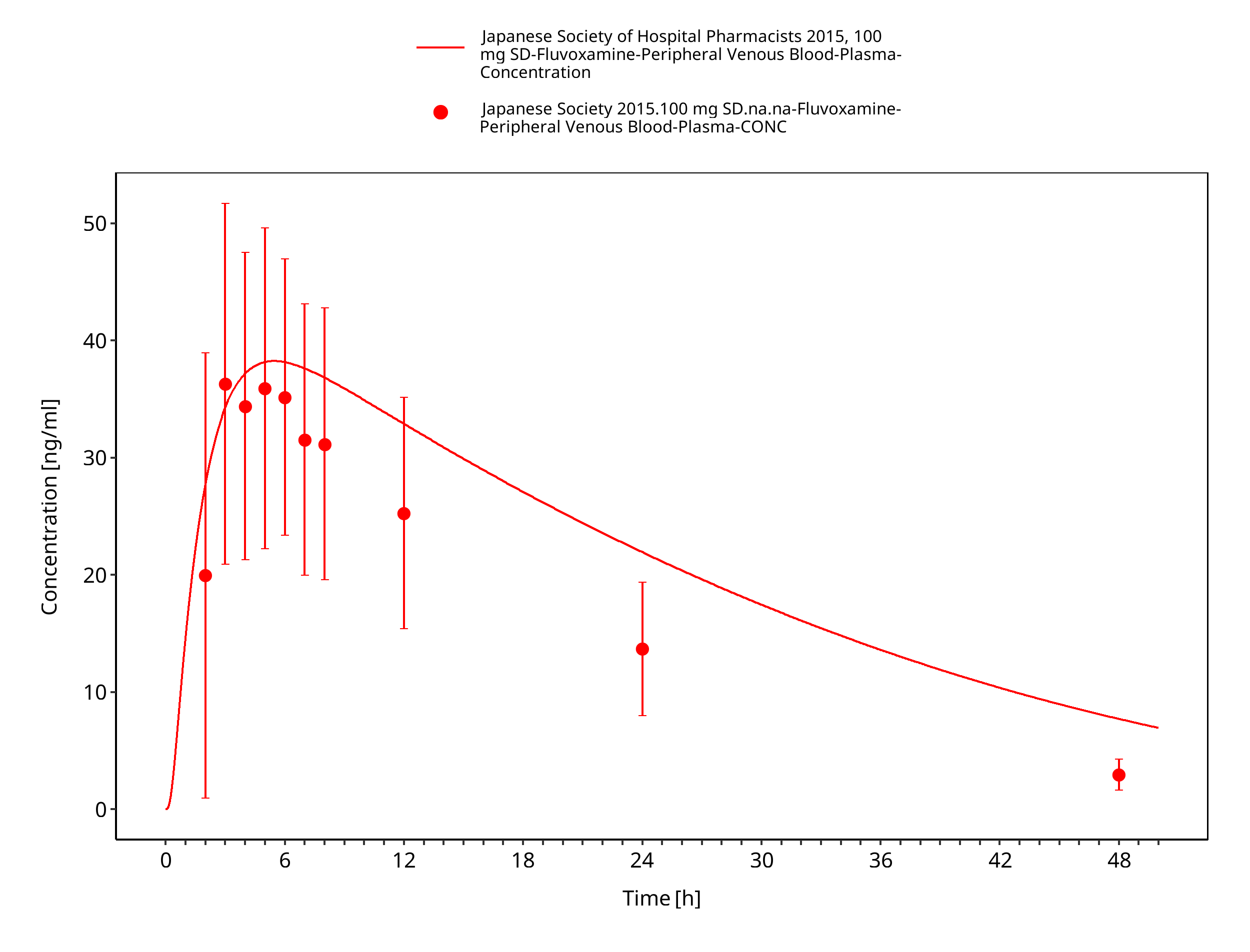
Figure 3-21: Time Profile Analysis
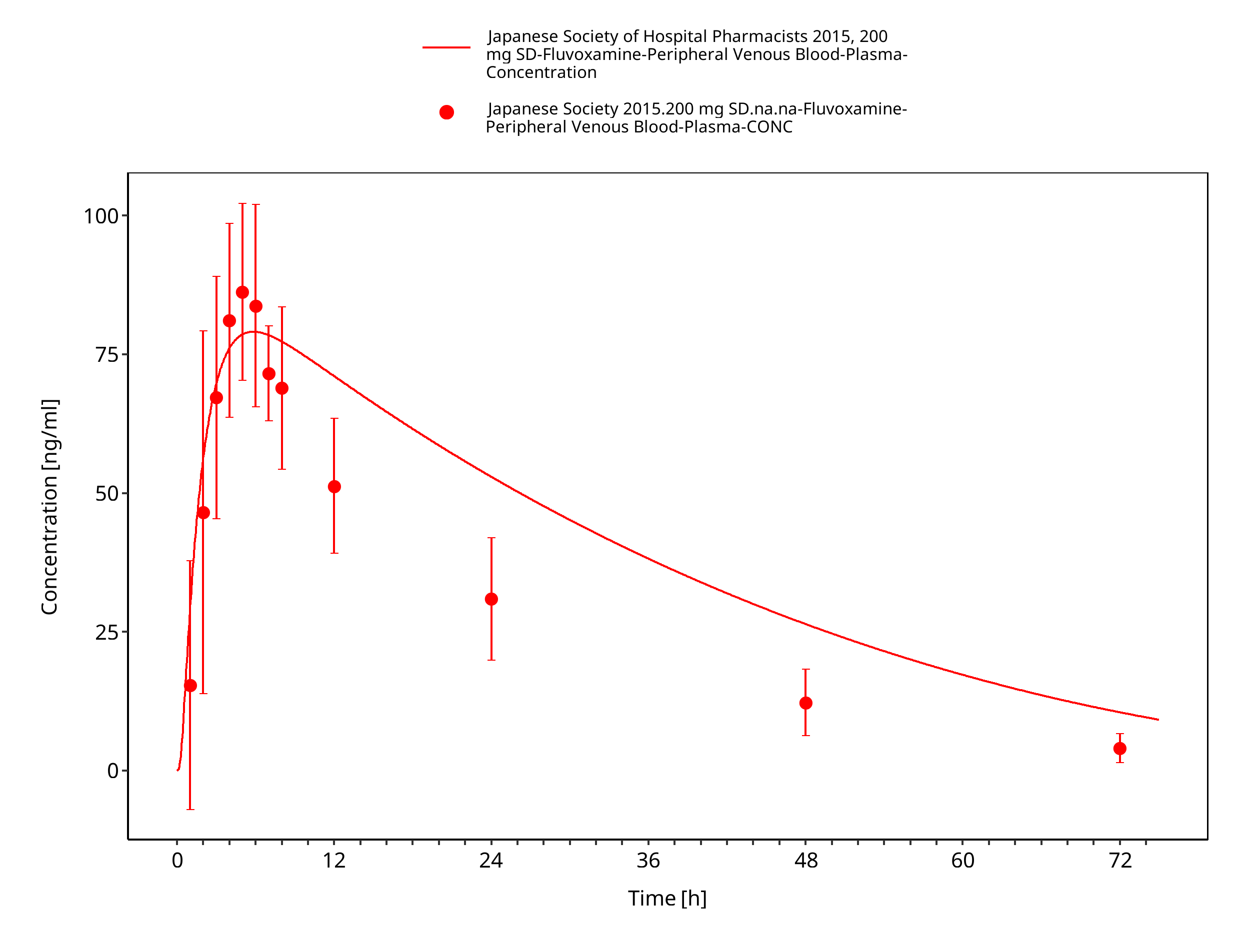
Figure 3-22: Time Profile Analysis
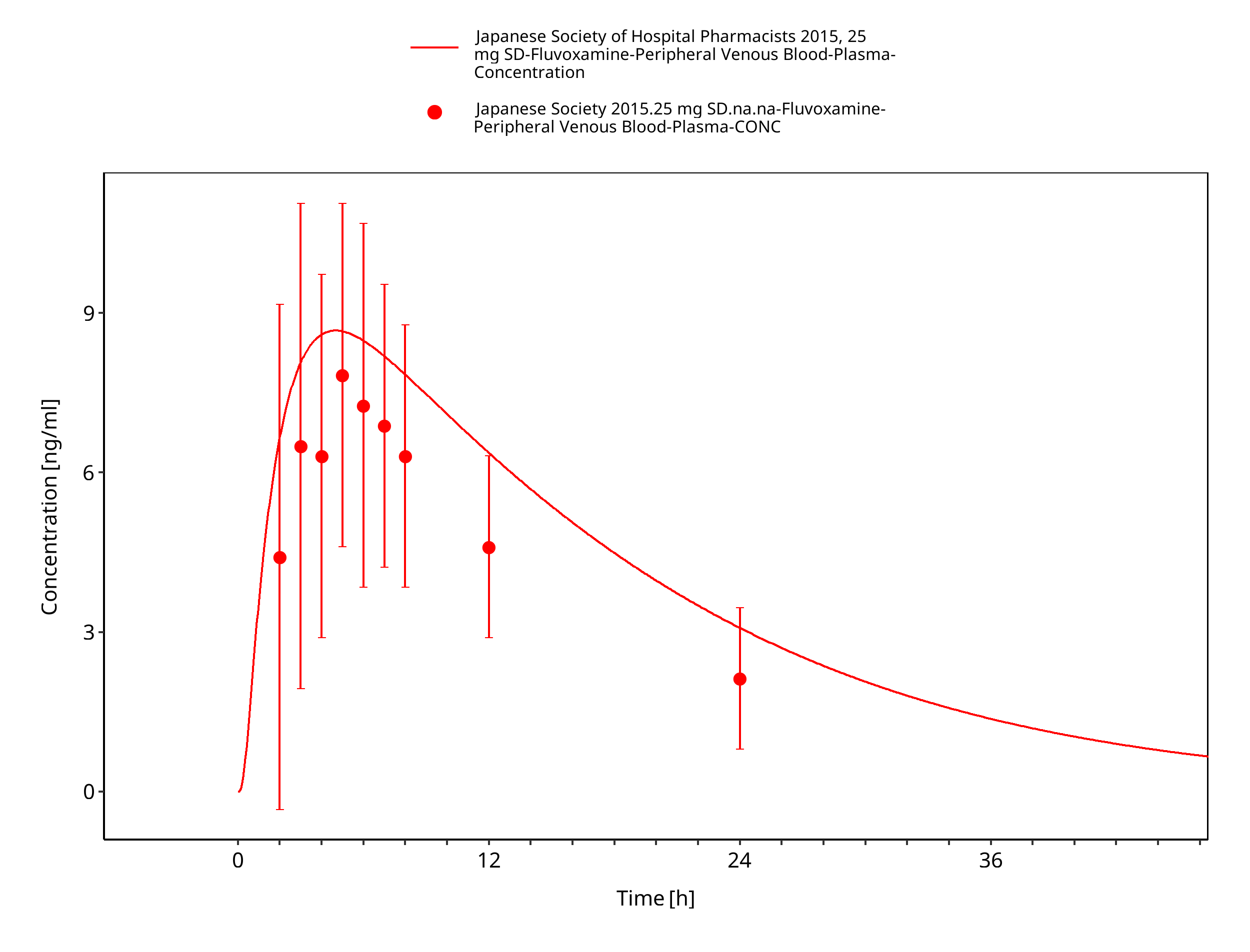
Figure 3-23: Time Profile Analysis
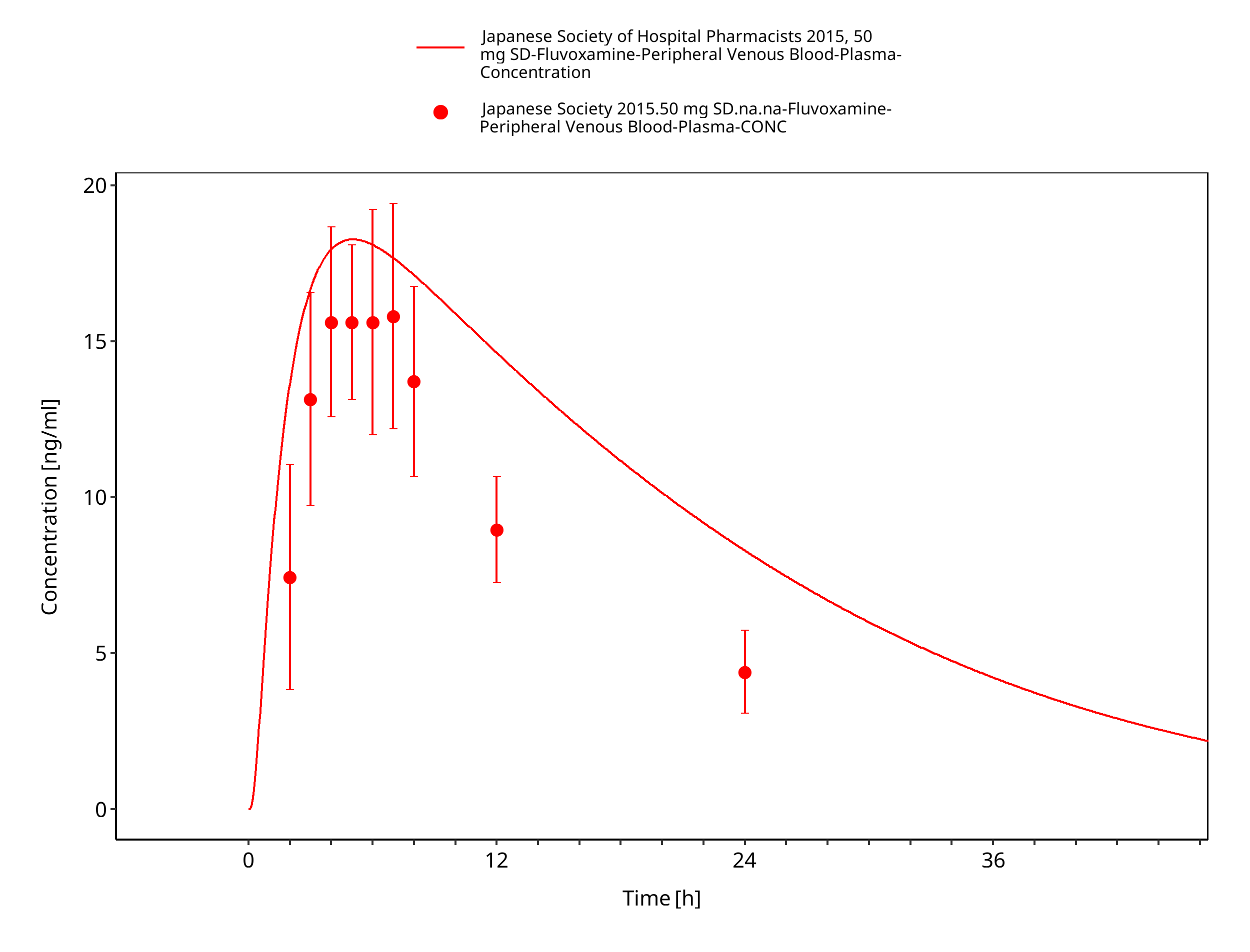
Figure 3-24: Time Profile Analysis
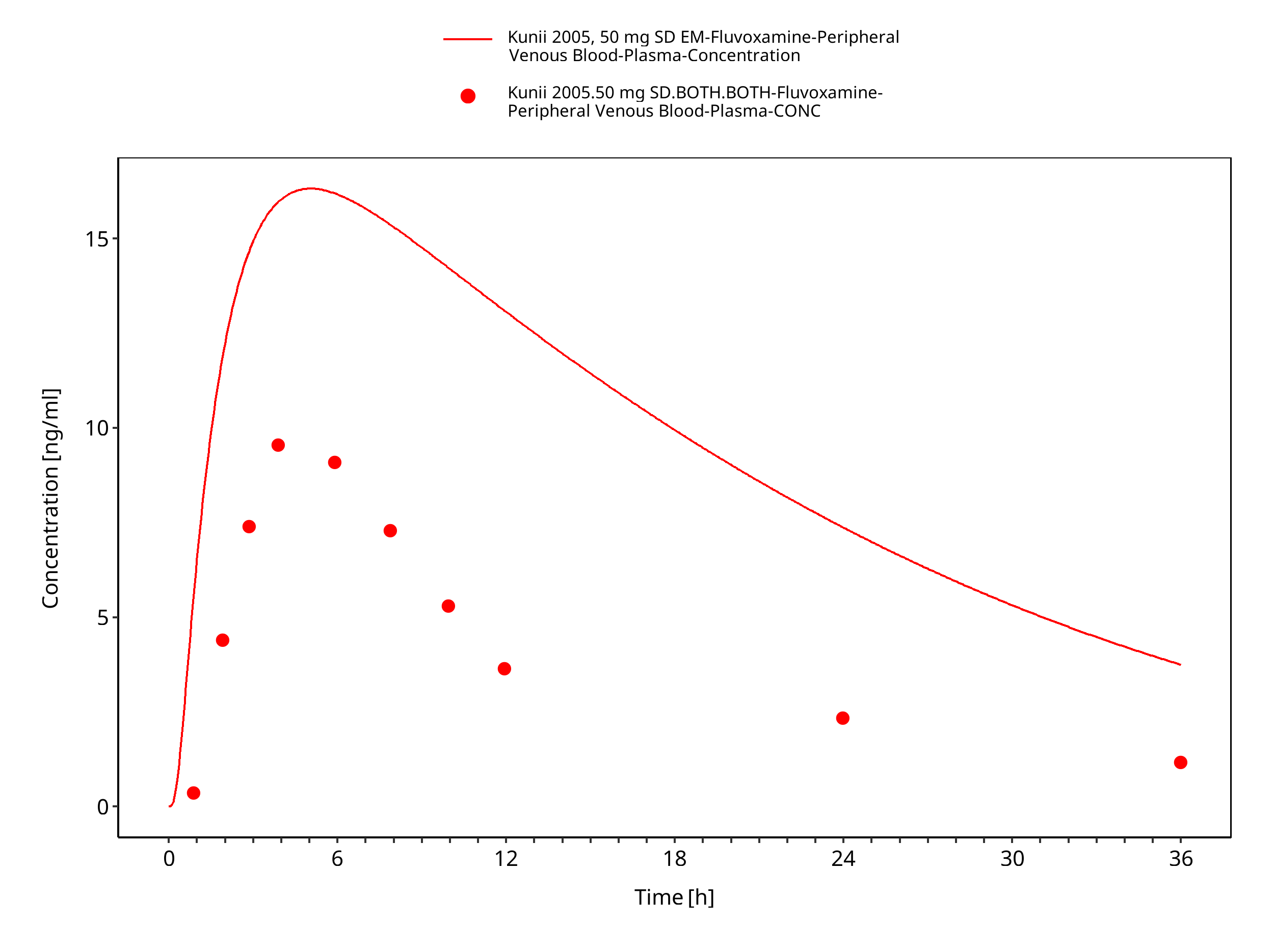
Figure 3-25: Time Profile Analysis
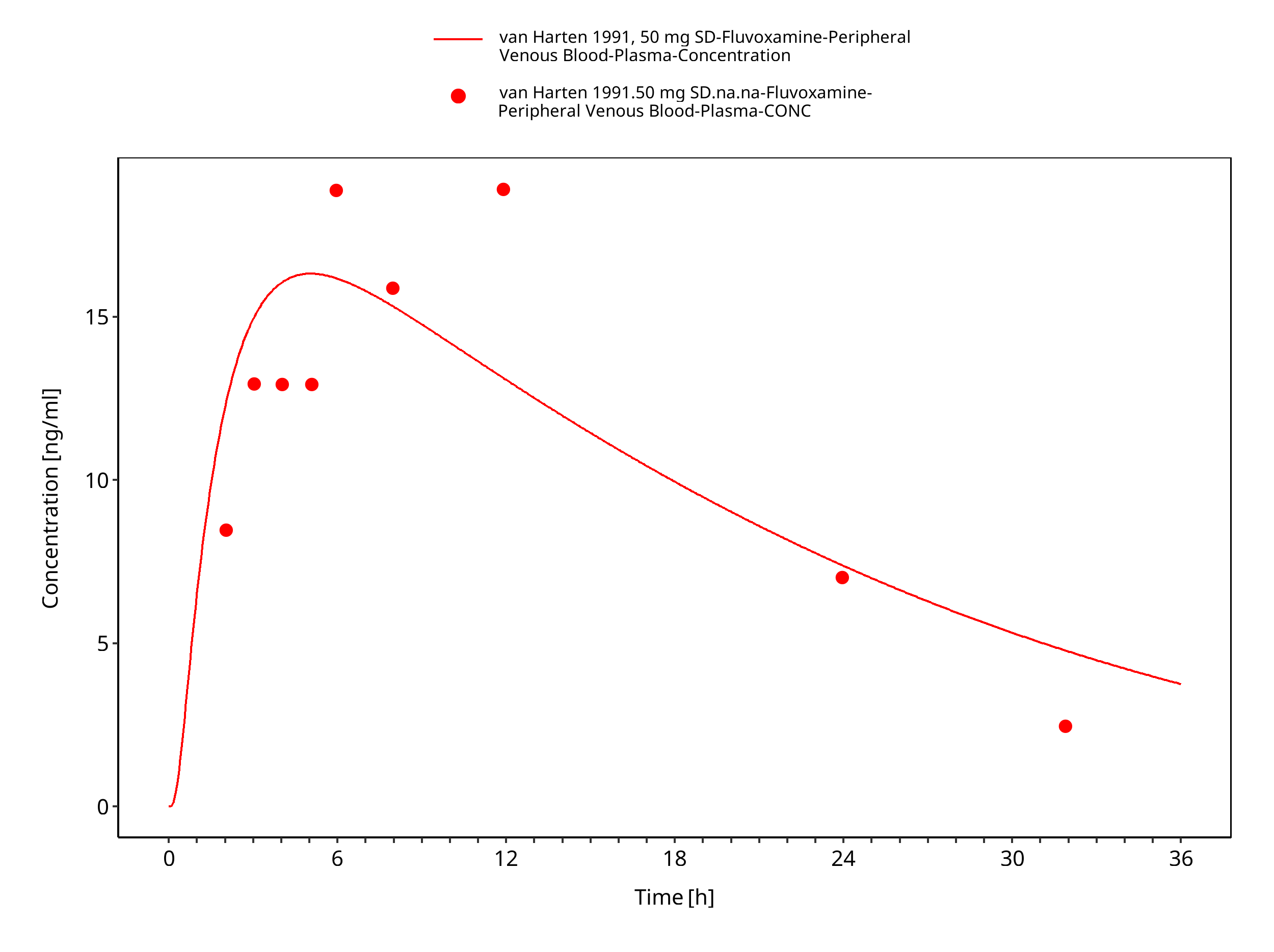
Figure 3-26: Time Profile Analysis
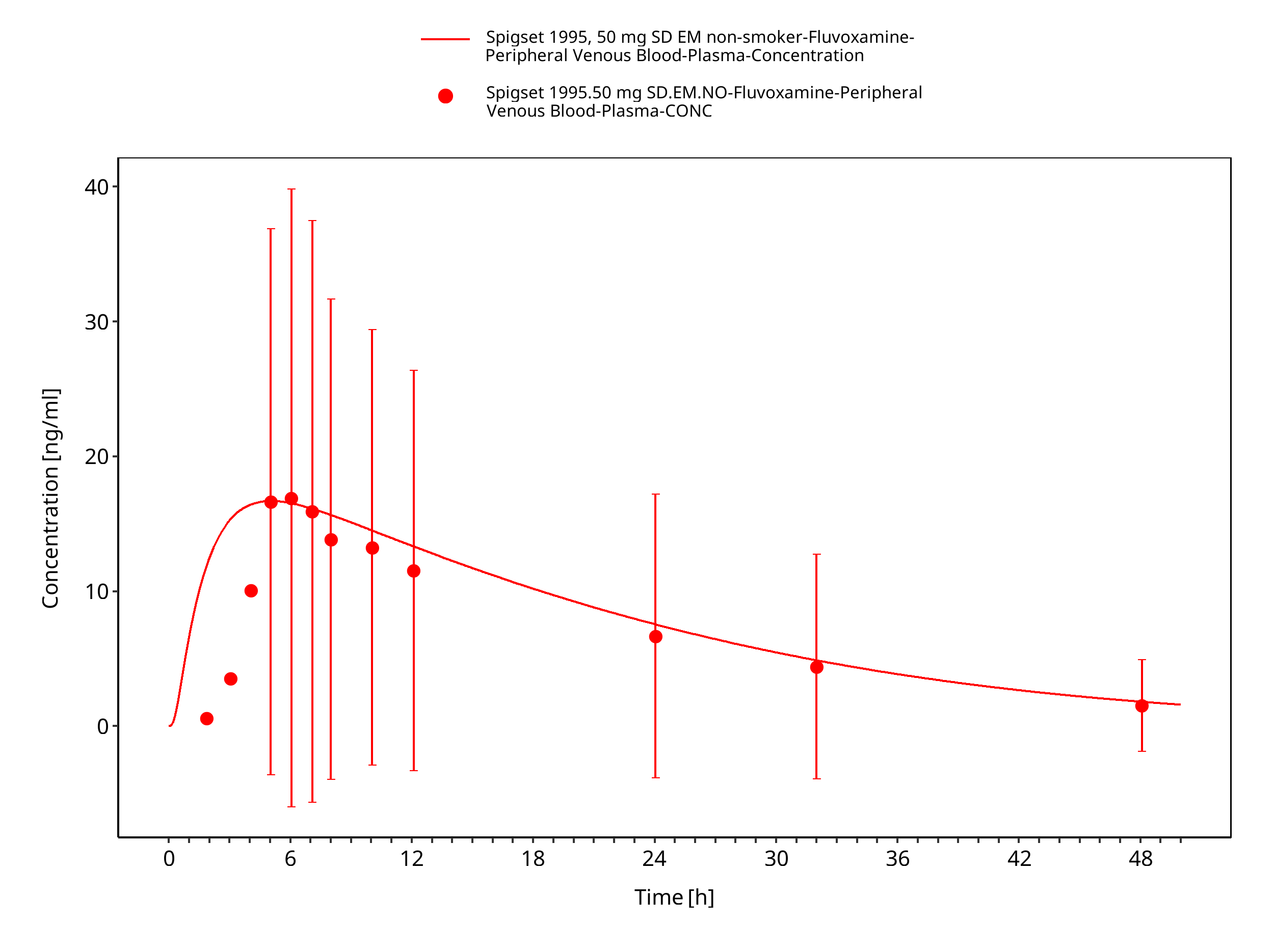
Figure 3-27: Time Profile Analysis
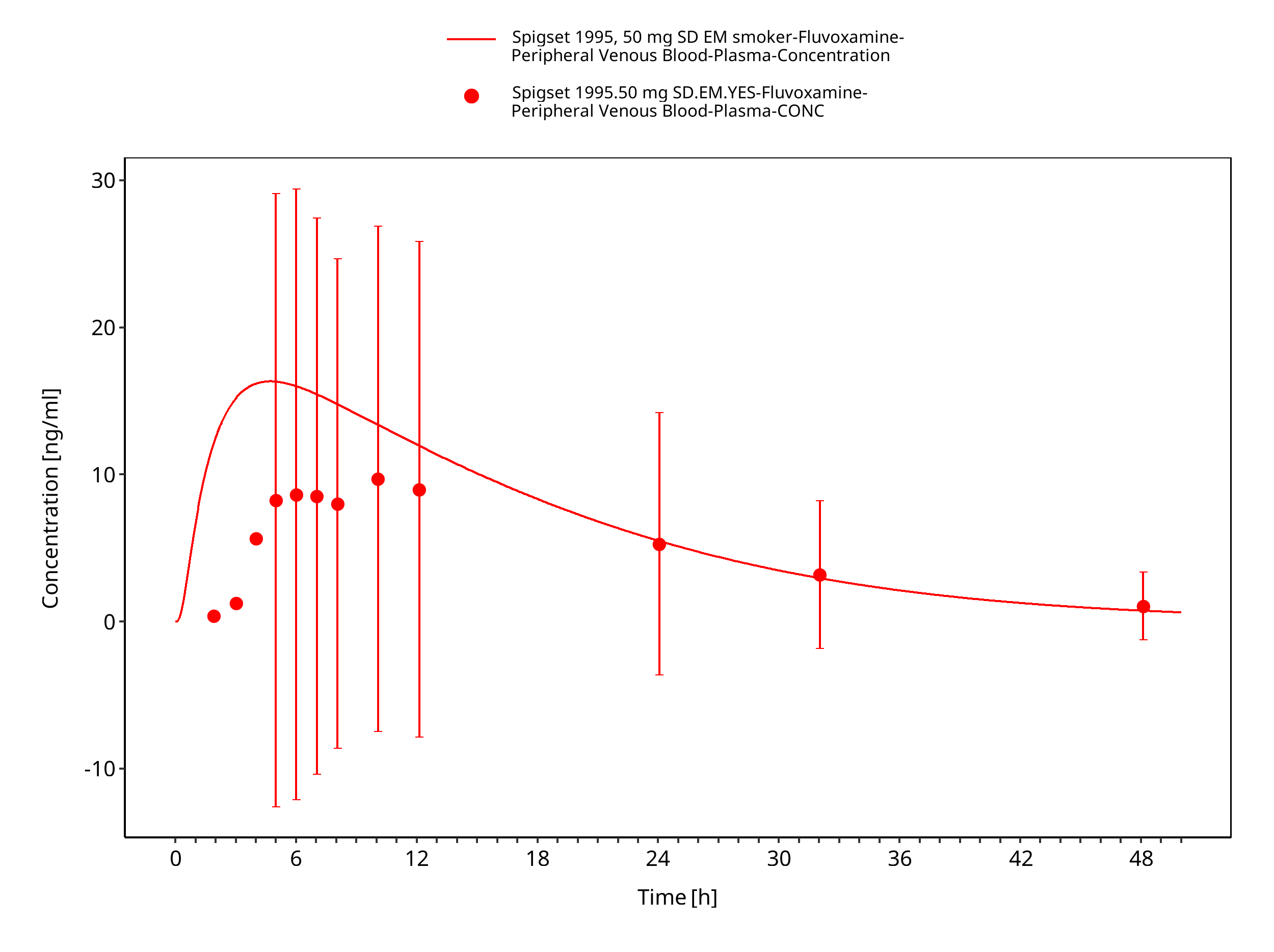
Figure 3-28: Time Profile Analysis
4 Conclusion¶
The herein presented PBPK model adequately describes the pharmacokinetics of fluvoxamine in adults.
In particular, it applies quantitative metabolism by CYP1A2 and CYP2D6. The inhibition of CYP1A2 and CYP3A4 are implemented and evaluated (shown elsewhere) in the current model as well. Thus, the model is fit for purpose to be applied for the prediction of drug-drug interaction
5 References¶
ANI Pharmaceuticals Inc. 2008 ANI Pharmaceuticals Inc. Fluvoxamine maleate - prescribing information. (2008).
Bahrami 2007 Bahrami, G. & Mohammadi, B. Rapid and sensitive bioanalytical method for measurement of fluvoxamine in human serum using 4-chloro-7-nitrobenzofurazan as pre-column derivatization agent: application to a human pharmacokinetic study. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 857, 322–6 (2007).
Britz 2019 Physiologically-based pharmacokinetic models for CYP1A2 drug–drug interaction prediction: a modeling network of fluvoxamine, theophylline, caffeine, rifampicin, and midazolam. CPT Pharmacometrics Syst. Pharmacol. 8, 296-307 (2019)
Christensen 2002 Christensen, M. et al. Low daily 10-mg and 20-mg doses of fluvoxamine inhibit the metabolism of both caffeine (cytochrome P4501A2) and omeprazole (cytochrome P4502C19). Clin. Pharmacol. Ther. 71, 141–52 (2002).
Claassen 1983 Claassen, V. Review of the animal pharmacology and pharmacokinetics of fluvoxamine. Br. J. Clin. Pharmacol. 15, 349S–355S (1983).
Crews 2014 Crews, K.R. et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin. Pharmacol. Ther. 95, 376–82 (2014).
DeBree 1983 DeBree, H., VanderSchoot, J. & Post, L. Fluvoxamine maleate; Disposition in man. Eur. J. Drug Metab. Pharmacokinet. 8, 175–79 (1983).
DeVries 1992 DeVries, M., VanHarten, J., VanBemmel, P. & Raghoebar, M. Single and multiple oral dose fluvoxamine kinetics in young and elderly subjects. Ther. Drug Monit. 14, 493–98 (1992).
Drugbank (https://www.drugbank.ca/drugs/DB00176), last view: 22 October 2018;
Fleishaker 1994 Fleishaker, J. & Hulst, L. A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine. Eur. J. Clin. Pharmacol. 46, 35–9 (1994).
Fukasawa 2006 Fukasawa, T. et al. Effects of caffeine on the kinetics of fluvoxamine and its major metabolite in plasma after a single oral dose of the drug. Ther. Drug Monit. 28, 308–11 (2006).
Hallifax 2007 Hallifax, D. & Houston, J.B. Saturable uptake of lipophilic amine drugs into isolated hepatocytes: mechanisms and consequences for quantitative clearance prediction. Drug Metab. Dispos. 35, 1325–32 (2007).
Japanese Society 2015 Japanese Society of Hospital Pharmacists. 医薬品インタビューフォーム. (2015).
Karjalainen 2008 Karjalainen, M.J., Neuvonen, P.J. & Backman, J.T. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin. Pharmacol. Toxicol. 103, 157–65 (2008).
Kuepfer 2016 Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Block M, Eissing T, Teutonico D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model.CPT Pharmacometrics Syst Pharmacol. 2016 Oct;5(10):516-531. doi: 10.1002/psp4.12134. Epub 2016 Oct 19.
Kunii 2005 Kunii, T. et al. Interaction study between enoxacin and fluvoxamine. Ther. Drug Monit. 27, 349–53 (2005).
Labellarte 2004 Labellarte, M. et al. Multiple-dose pharmacokinetics of fluvoxamine in children and adolescents. J. Am. Acad. Child Adolesc. Psychiatry 43, 1497–505 (2004).
Meyer 2012 Meyer, M., Schneckener, S., Ludewig, B., Kuepfer, L. & Lippert, J. Using expression data for quantification of active processes in physiologically-based pharmacokinetic modeling. Drug Metab. Dispos. 40, 892–901 (2012).
Miura 2007 Miura, M. & Ohkubo, T. Identification of human cytochrome P450 enzymes involved in the major metabolic pathway of fluvoxamine. Xenobiotica. 37, 169–79 (2007).
MSDS material safety data sheet of fluvoxamine
Olesen 2000 Olesen, O.V. & Linnet, K. Fluvoxamine-Clozapine drug interaction: inhibition in vitro of five cytochrome P450 isoforms involved in clozapine metabolism. J. Clin. Psychopharmacol. 20, 35–42 (2000).
Orlando 2010 Orlando, R., DeMartin, S., Andrighetto, L., Floreani, M. & Palatini, P. Fluvoxamine pharmacokinetics in healthy elderly subjects and elderly patients with chronic heart failure. Br. J. Clin. Pharmacol. 69, 279–86 (2010).
Perucca 1994 Perucca, E., Gatti, G. & Spina, E. Clinical pharmacokinetics of fluvoxamine. Clin. Pharmacokinet. 27, 175–90 (1994).
PK-Sim Ontogeny Database Version 7.3 (https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/38cf71b384cfc25cfa0ce4d2f3addfd32757e13b/PK-Sim%20Ontogeny%20Database%20Version%207.3.pdf)
Schlender 2016 Schlender JF, Meyer M, Thelen K, Krauss M, Willmann S, Eissing T, Jaehde U. Development of a Whole-Body Physiologically Based Pharmacokinetic Approach to Assess the Pharmacokinetics of Drugs in Elderly Individuals. Clin Pharmacokinet. 2016 Dec;55(12):1573-1589.
Spigset 1995 Spigset, O., Carleborg, L., Hedenmalm, K. & Dahlqvist, R. Effect of cigarette smoking on fluvoxamine pharmacokinetics in humans. Clin. Pharmacol. Ther. 58, 399–403 (1995).
Spigset 1997 Spigset, O., Granberg, K., Hägg, S., Norström, A. & Dahlqvist, R. Relationship between fluvoxamine pharmacokinetics and CYP2D6/CYP2C19 phenotype polymorphisms. Eur. J. Clin. Pharmacol. 52, 129–33 (1997).
Spigset 1998 Spigset, O., Granberg, K., Hägg, S., Söderström, E. & Dahlqvist, R. Non-linear fluvoxamine disposition. Br. J. Clin. Pharmacol. 45, 257–63 (1998).
Spigset 2001 Spigset, O., Axelsson, S., Norström, A., Hägg, S. & Dahlqvist, R. The major fluvoxamine metabolite in urine is formed by CYP2D6. Eur. J. Clin. Pharmacol. 57, 653–8 (2001).
FDA 2017 U.S. Food and Drug Administration. Clinical Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Draft Guidance for Industry. (2017).
VanHarten 1991 VanHarten, J., VanBemmel, P., Dobrinska, M.R., Ferguson, R.K. & Raghoebar, M. Bioavailability of fluvoxamine given with and without food. Biopharm. Drug Dispos. 12, 571–6 (1991).
Yao 2001 Yao, C. et al. Fluvoxamine-theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin. Pharmacol. Ther. 70, 415–24 (2001).
Zhou 2009 Zhou, S.F., Yang, L.P., Zhou, Z.W., Liu, Y.H. & Chan, E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 11, 481–494 (2009).
Related Models¶
Explore other Other Compounds models in this library:
- Moclobemide - PBPK model and evaluation report
- Tizanidine - PBPK model and evaluation report