Building and evaluation of a PBPK model for Felodipine in healthy adults¶
| Version | 2.0-OSP12.3 |
|---|---|
| based on Model Snapshot and Evaluation Plan | https://github.com/Open-Systems-Pharmacology/Felodipine-Model/releases/tag/v2.0 |
| OSP Version | 12.3 |
| Qualification Framework Version | 3.7 |
This evaluation report and the corresponding PK-Sim project file are filed at:
https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/
Table of Contents¶
- 1 Introduction
- 2 Methods
- 2.1 Modeling Strategy
- 2.2 Data
- 2.3 Model Parameters and Assumptions
- 3 Results and Discussion
- 3.1 Final input parameters
- 3.2 Diagnostics Plots
- 3.3 Concentration-Time Profiles
- 4 Conclusion
- 5 References
1 Introduction¶
Felodipine is a calcium-channel blocker, indicated for angina pectoris and arterial hypertension. It is mostly metabolized by CYP3A4 making it a sensitive probe and victim drug for the investigation of CYP3A4 activity in vivo. It is a BCS class II compound. Felodipine shows substantial first pass metabolism resulting in a bioavailability of 15%.
The model has been developed and evaluated by comparing observed data to simulations of a large number of clinical studies covering a dose range of 1.5 mg to 10 mg after intravenous and oral administrations. Furthermore, it has been evaluated within a CYP3A4 DDI modeling network as a victim drug.
Model features include:
- metabolism by CYP3A4
- metabolism by an unknown enzyme via unspecific hepatic clearance
- a decrease in the permeability between the intracellular and interstitial space (model parameters
P (intracellular->interstitial)andP (interstitial->intracellular)) in intestinal mucosa to optimize quantitatively the extent of gut wall metabolism
2 Methods¶
2.1 Modeling Strategy¶
The general concept of building a PBPK model has previously been described by Kuepfer et al. (Kuepfer 2016). Relevant information on anthropometric (height, weight) and physiological parameters (e.g. blood flows, organ volumes, binding protein concentrations, hematocrit, cardiac output) in adults was gathered from the literature and has been previously published (Willmann 2007). The information was incorporated into PK-Sim® and was used as default values for the simulations in adults.
The applied activity and variability of plasma proteins and active processes that are integrated into PK-Sim® are described in the publicly available PK-Sim® Ontogeny Database Version 7.3 (PK-Sim Ontogeny Database Version 7.3) or otherwise referenced for the specific process.
First, a mean model was built using clinical data from single dose and multiple doses studies with intravenous and oral administration of felodipine (Edgar 1987, Bailey 1996, Gelal 2005, Bailey 2003, Goosen 2004, Jalava 1997, Blychert 1990, Lundahl 1998, Bailey 1995, Aberg 1997, Bailey 1993, Edgar 1992, Lundahl 1997). One DDI study (Jalava 1997) was also used in the optimization to help the model describe DDI better. The mean PBPK model was developed using a typical male European individual. The relative tissue-specific expressions of enzymes predominantly being involved in the metabolism of felodipine (CYP3A4) were considered (Meyer 2012).
A specific selected set of parameters (see below) was optimized using the Parameter Identification module provided in PK-Sim®. Structural model selection was mainly guided by visual inspection of the resulting description of data and biological plausibility.
Once the appropriate structural model was identified, dissolution kinetic parameters were optimized for immediate-release tablets.
The model was then evaluated by simulating further clinical studies reporting pharmacokinetic concentration-time profiles of felodipine.
Details about input data (physicochemical, in vitro and clinical) can be found in Section 2.2.
Details about the structural model and its parameters can be found in Section 2.3.
2.2 Data¶
2.2.1 In vitro and physicochemical data¶
A literature search was performed to collect available information on physicochemical properties of felodipine. The obtained information from literature is summarized in the table below, and is used for model building.
| Parameter | Unit | Value | Source | Description |
|---|---|---|---|---|
| MW | g/mol | 384.254 | DrugBank DB01023 | Molecular weight |
| pKa1 | n.a. | Alskar 2018 | Acid dissociation constant of conjugate acid | |
| pKa1 | 5.07 | Pandey 2013 | Acid dissociation constant of conjugate acid; compound type: acid | |
| Solubility (pH) | mg/L | 14.3 (7.1) |
Takano 2016 | Aqueous Solubility |
| 1 | Scholz 2002 | Aqueous Solubility | ||
| 53 | Söderlind 2010 | Solubility in fasted state simulated intestinal fluid I | ||
| 12 | Söderlind 2010 | Solubility in fasted state simulated intestinal fluid II | ||
| 14 | Söderlind 2010 | Solubility in fasted human intestinal fluid | ||
| 15 (7.5) |
Persson 2005 | Solubility in fasted human intestinal fluid | ||
| 413 (6.1) |
Persson 2005 | Solubility in fed human intestinal fluid | ||
| 191 | Persson 2005 | Solubility in fed state simulated intestinal fluid | ||
| 77 (6.35) |
Scholz 2002 | Solubility in fasted state chyme | ||
| 56 (4.93) |
Scholz 2002 | Solubility in fed state chyme | ||
| logP | 4.36 | DrugBank DB01023 | Partition coefficient between octanol and water | |
| 3.44 | DrugBank DB01023 | Partition coefficient between octanol and water | ||
| 3.86 | McPherson 2020 | Partition coefficient between octanol and water | ||
| 4.5 | Scholz 2002 | Partition coefficient between octanol and water | ||
| 4.8 | Bu 2006 | Partition coefficient between octanol and water | ||
| fu | % | 0.36 | Soons 1993 | Fraction unbound in plasma |
| % | 0.36 | Ushimura 2010 | Fraction unbound in plasma | |
| Vmax, Km CYP3A | pmol/mg/min, µmol/L |
1630 2.81 |
Walsky 2004 | CYP3A liver microsomes Michaelis-Menten kinetics |
| Vmax, Km CYP3A | pmol/mg/min, µmol/L |
240 6.9 |
Bu 2006 | CYP3A liver microsomes Michaelis-Menten kinetics |
| Vmax, Km CYP3A4 | pmol/mg/min, µmol/L |
36.8 0.938 |
Walsky 2004 | Recombinant CYP3A4 Michaelis-Menten kinetics |
| Vmax, Km CYP3A5 | pmol/mg/min, µmol/L |
24.2 1.41 |
Walsky 2004 | Recombinant CYP3A5 Michaelis-Menten kinetics |
2.2.2 Clinical data¶
A literature search was performed to collect available clinical data on felodipine in adults.
The following publications were found in adults for model building:
| Publication | Arm / Treatment / Information used for model building |
|---|---|
| Lundahl 1997 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 1.5 mg intravenous infusion |
| Edgar 1987 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine: - 10 mg oral solution - 10 mg extended release tablet - 10 mg immediate release tablet |
| Blychert 1990 | Plasma PK profiles in healthy subjects with multiple dose administrations of a felodipine: - 10 mg oral solution - 10 mg extended release tablet |
| Goosen 2004 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 5 mg extended release tablet |
| Jalava 1997 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 5 mg extended release tablet - alone (control) - with itraconazole (treatment) |
| Bailey 1996 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 10 mg extended release tablet |
| Gelal 2005 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 10 mg extended release tablet |
| Bailey 2003 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 10 mg extended release tablet |
| Bailey 1993 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 5 mg immediate release tablet |
| Edgar 1992 | Plasma PK profiles in healthy subjects with single dose administrations of a felodipine 5 mg immediate release tablet |
| Blychert 1990 | Plasma PK profiles in healthy subjects with multiple dose administrations of a felodipine: - 10 mg oral solution - 10 mg extended release tablet |
| Lundahl 1998 | Plasma PK profiles in healthy subjects with multiple dose administrations of a felodipine 10 mg extended release tablet |
| Bailey 1995 | Plasma PK profiles in healthy subjects with multiple dose administrations of a felodipine 10 mg extended release tablet |
| Aberg 1997 | Plasma PK profiles in healthy subjects with multiple dose administrations of a felodipine 10 mg extended release tablet |
The following dosing scenarios were simulated and compared to respective data for model verification:
| Scenario | Data reference |
|---|---|
| po 5 mg single dose (extended release tablet) | Dresser 2000 |
| po 10 mg single dose (extended release tablet) | Dresser 2017 |
| Dresser 2002 | |
| Bailey 2000 | |
| Bailey 1998 | |
| Lundahl 1997 | |
| Madsen 1996 | |
| po 2.5 / 5 mg once daily (extended release tablet) | Dresser 2000 |
| po 10 mg once daily (immediate release tablet) | Blychert 1990 |
| po 10 mg twice daily (immediate release tablet) | Blychert 1990 |
| Smith 1987 | |
| po 10 mg three times daily (immediate release tablet) | Bratel 1989 |
2.3 Model Parameters and Assumptions¶
2.3.1 Absorption¶
The model parameter Specific intestinal permeability was was calculated by PK-Sim® and kept to that value since it was insensitive. The default solubility was assumed to be measured value in fasted state simulated intestinal fluid (see Section 2.2.1)
The dissolution of both immediate and extended-release tables was implemented via two Weibull dissolution tablets, and the dissolution kinetic parameters were optimized (see Section 2.3.4).
2.3.2 Distribution¶
Felodipine is highly bound to proteins in plasma (see Section 2.2.1). A value of 0.36% was used in this PBPK model for Fraction unbound (plasma, reference value).
An important parameter influencing the resulting volume of distribution is lipophilicity. The reported experimental logP values are in the range of 4 (see Section 2.2.1) which served as a starting value. Finally, the model parameter Lipophilicity was optimized to match best clinical data (see also Section 2.3.4).
After testing the available organ-plasma partition coefficient and cell permeability calculation methods built in PK-Sim, observed clinical data was best described by choosing the partition coefficient calculation by Rodgers and Rowland and cellular permeability calculation by PK-Sim Standard.
2.3.3 Metabolism and Elimination¶
Two metabolic pathways were implement into the model via Michaelis-Menten kinetics
- CYP3A4
- unknown hepatic enzyme via unspecific hepatic clearance
The latter was preferred over renal clearance, since there is evidence that felodipine is fully metabolized and not found in urine (Edgar 1987). CYP3A5 was not implemented since the fraction metabolized appeared to be minor compared to CYP3A4.
The CYP3A4 expression profiles is based on high-sensitive real-time RT-PCR (Nishimura 2003). Absolute tissue-specific expressions were obtained by considering the respective absolute concentration in the liver. The PK-Sim database provides a default value for CYP3A4 (compare Rodrigues 1999 and assume 40 mg protein per gram liver).
The first model simulations showed that gut wall metabolism was underrepresented in the PBPK model. In order to increase gut wall metabolism, the “mucosa permeability on basolateral side” (jointly the model parameters in the mucosa: P (interstitial->intracellular) and P (intracellular->interstitial)) was estimated. A decrease in this permeability may lead to higher gut wall concentrations and, in turn, to a higher gut wall elimination. This parameter was preferred over other parameters such as relative CYP3A4 expression or fraction unbound (fu) in the gut wall as it is technically not limited to a maximum value of 100%.
2.3.4 Automated Parameter Identification¶
This is the result of the final parameter identification for the base model:
| Model Parameter | Optimized Value | Unit |
|---|---|---|
Lipophilicity |
4.51 | Log Units |
| Basolateral mucosa permeability ( P (interstitial->intracellular), P (intracellular->interstitial)) |
0.09 | cm/min |
kcat (CYP3A4) |
204.70 | 1/min |
Dissolution time (extended release tablet) |
286.95 | min |
Dissolution shape (extended release tablet) |
0.76 | |
Lag time (extended release tablet) |
18.68 | min |
Tablet time delay factor (extended release tablet) |
0.07 | |
Dissolution time (immediate release tablet) |
46.50 | min |
Dissolution shape (immediate release tablet) |
0.89 |
3 Results and Discussion¶
The PBPK model for felodipine was developed and verified with clinical pharmacokinetic data.
The model was built and evaluated covering data from studies including in particular
- intravenous (infusions) and oral administrations (solutions, immediate release and extended release tablets).
- a dose range of 1.5 to 10 mg.
The model quantifies metabolism via CYP3A4, and a second unknown hepatic enzyme.
The next sections show:
- the final model input parameters for the building blocks: Section 3.1.
- the overall goodness of fit: Section 3.2.
- simulated vs. observed concentration-time profiles for the clinical studies used for model building and for model verification: Section 3.3.
3.1 Final input parameters¶
The compound parameter values of the final PBPK model are illustrated below.
Compound: Felodipine¶
Parameters¶
| Name | Value | Value Origin | Alternative | Default |
|---|---|---|---|---|
| Solubility at reference pH | 12 µg/ml | Publication-Soderlind, 2010 | FaSSIF II | True |
| Reference pH | 6.5 | Publication-Soderlind, 2010 | FaSSIF II | True |
| Lipophilicity | 4.5085714616 Log Units | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:40 | Measurement | True |
| Fraction unbound (plasma, reference value) | 0.0036 | Publication-Ushimura, 2010 | Measurement | True |
| Cl | 2 | Internet-DrugBank DB01023 | ||
| Is small molecule | Yes | |||
| Molecular weight | 384.254 g/mol | Internet-DrugBank DB01023 | ||
| Plasma protein binding partner | Unknown |
Calculation methods¶
| Name | Value |
|---|---|
| Partition coefficients | Rodgers and Rowland |
| Cellular permeabilities | PK-Sim Standard |
Processes¶
Metabolizing Enzyme: CYP3A4-Walsky 2004¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 1630 pmol/min/mg mic. protein | Publication-Walsky 2004 |
| Km | 2.81 µmol/l | Publication-Walsky 2004 |
| kcat | 204.6995652687 1/min | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:40 |
Systemic Process: Total Hepatic Clearance-Unspecific hepatic clearance¶
Species: Human
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Fraction unbound (experiment) | 0.0036 | |
| Lipophilicity (experiment) | 4.3407865958 Log Units | |
| Plasma clearance | 0 ml/min/kg | |
| Specific clearance | 12.8042083376 1/min | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:41 |
Formulation: Felodipine_IR tablet¶
Type: Weibull
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Dissolution time (50% dissolved) | 46.5005948338 min | Parameter Identification-Parameter Identification-Value updated from 'PI IR Tablet alone' on 2022-08-10 11:29 |
| Lag time | 0 min | |
| Dissolution shape | 0.8876005929 | Parameter Identification-Parameter Identification-Value updated from 'PI IR Tablet alone' on 2022-08-10 11:29 |
| Use as suspension | Yes |
Formulation: Felodipine_ER tablet¶
Type: Weibull
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Dissolution time (50% dissolved) | 286.9463213309 min | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:41 |
| Lag time | 18.6758448616 min | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:41 |
| Dissolution shape | 0.7639975313 | Parameter Identification-Parameter Identification-Value updated from 'PI_IV+Solution+ER+DDI_additionalCL_3' on 2022-08-10 10:41 |
| Use as suspension | Yes |
3.2 Diagnostics Plots¶
Below you find the goodness-of-fit visual diagnostic plots for the PBPK model performance of all data used presented in Section 2.2.2.
The first plot shows observed versus simulated plasma concentration, the second weighted residuals versus time.
Table 3-1: GMFE for Felodipine concentration in plasma
| Group | GMFE |
|---|---|
| Intravenous administration (model building) | 1.33 |
| Oral administration, ER tablet (model building) | 1.30 |
| Oral administration, ER tablet (model verification) | 1.35 |
| Oral administration, IR tablet (model building) | 1.51 |
| Oral administration, IR tablet (model verification) | 1.35 |
| Oral administration, solution (model building) | 1.17 |
| All | 1.33 |
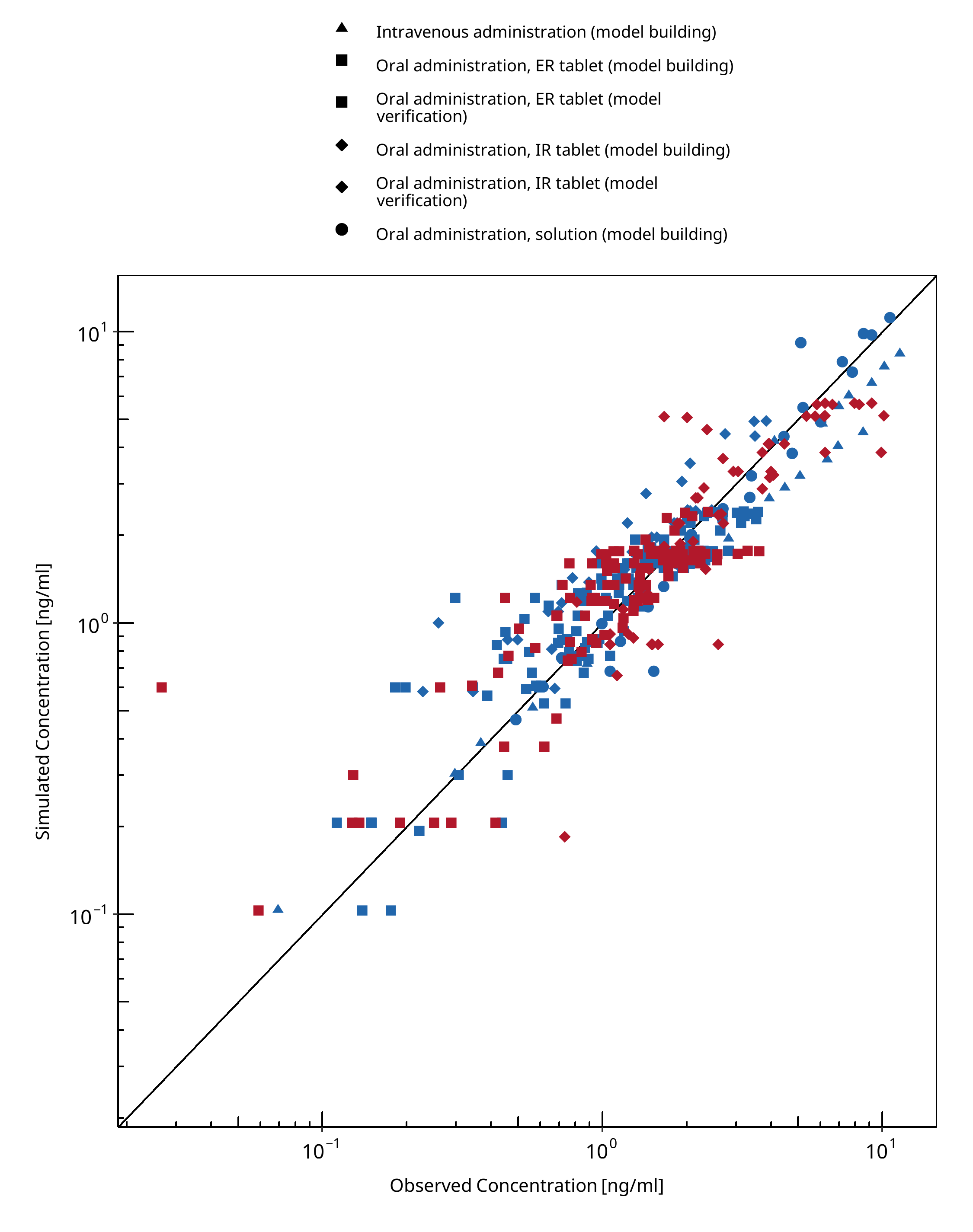
Figure 3-1: Felodipine concentration in plasma
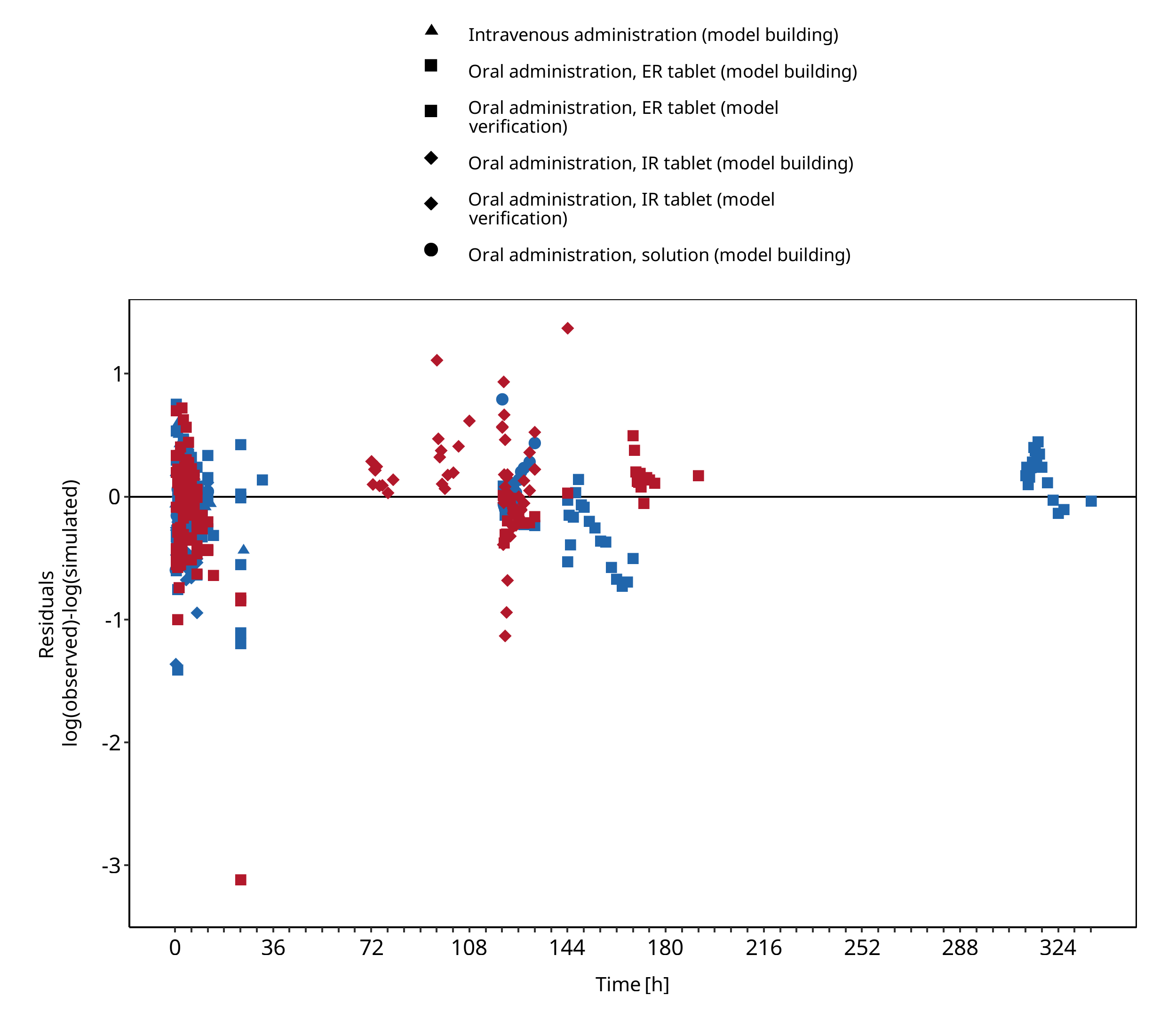
Figure 3-2: Felodipine concentration in plasma
3.3 Concentration-Time Profiles¶
Simulated versus observed concentration-time profiles of all data listed in Section 2.2.2 are presented below.
3.3.1 Model Building¶
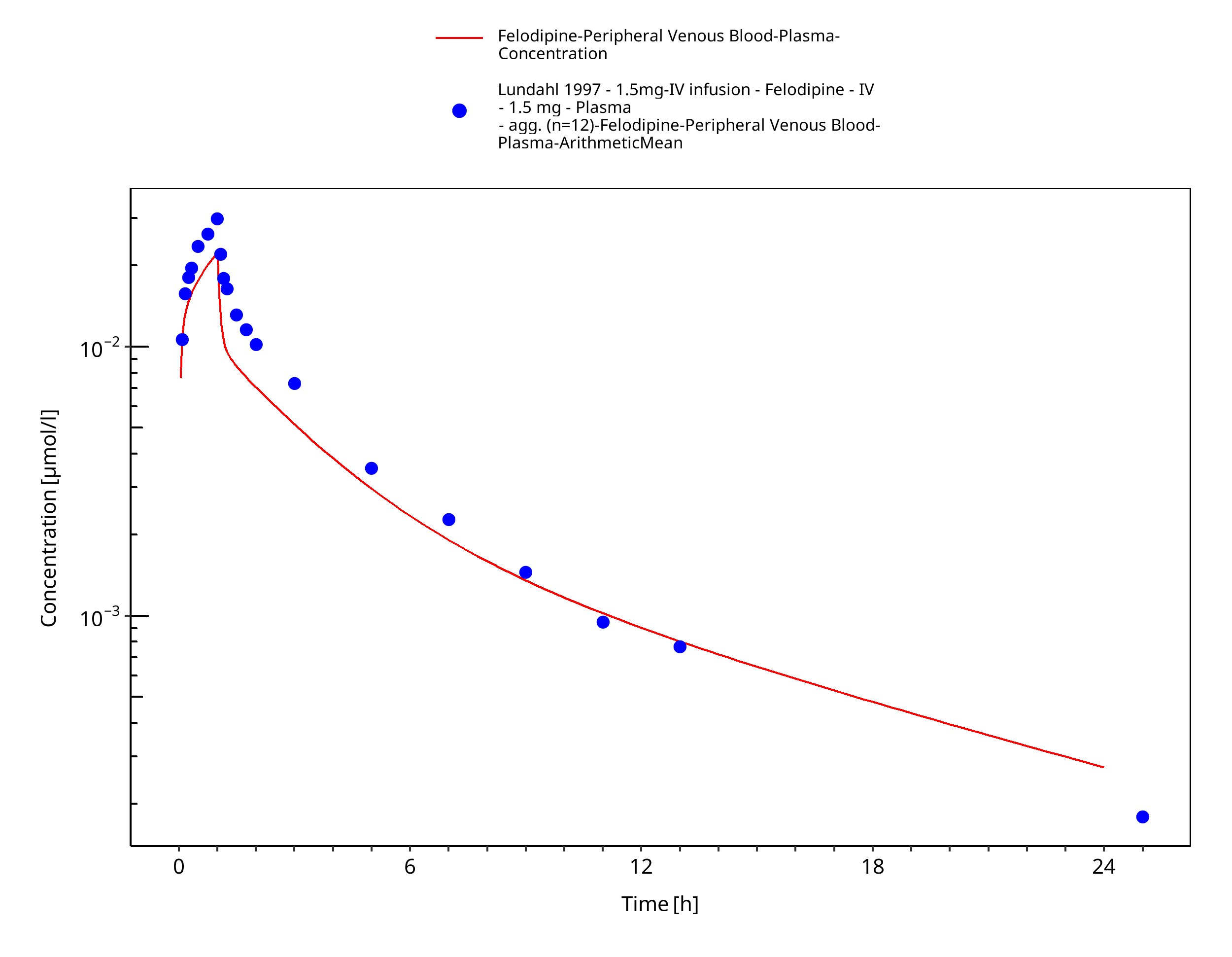
Figure 3-3: Time Profile Analysis
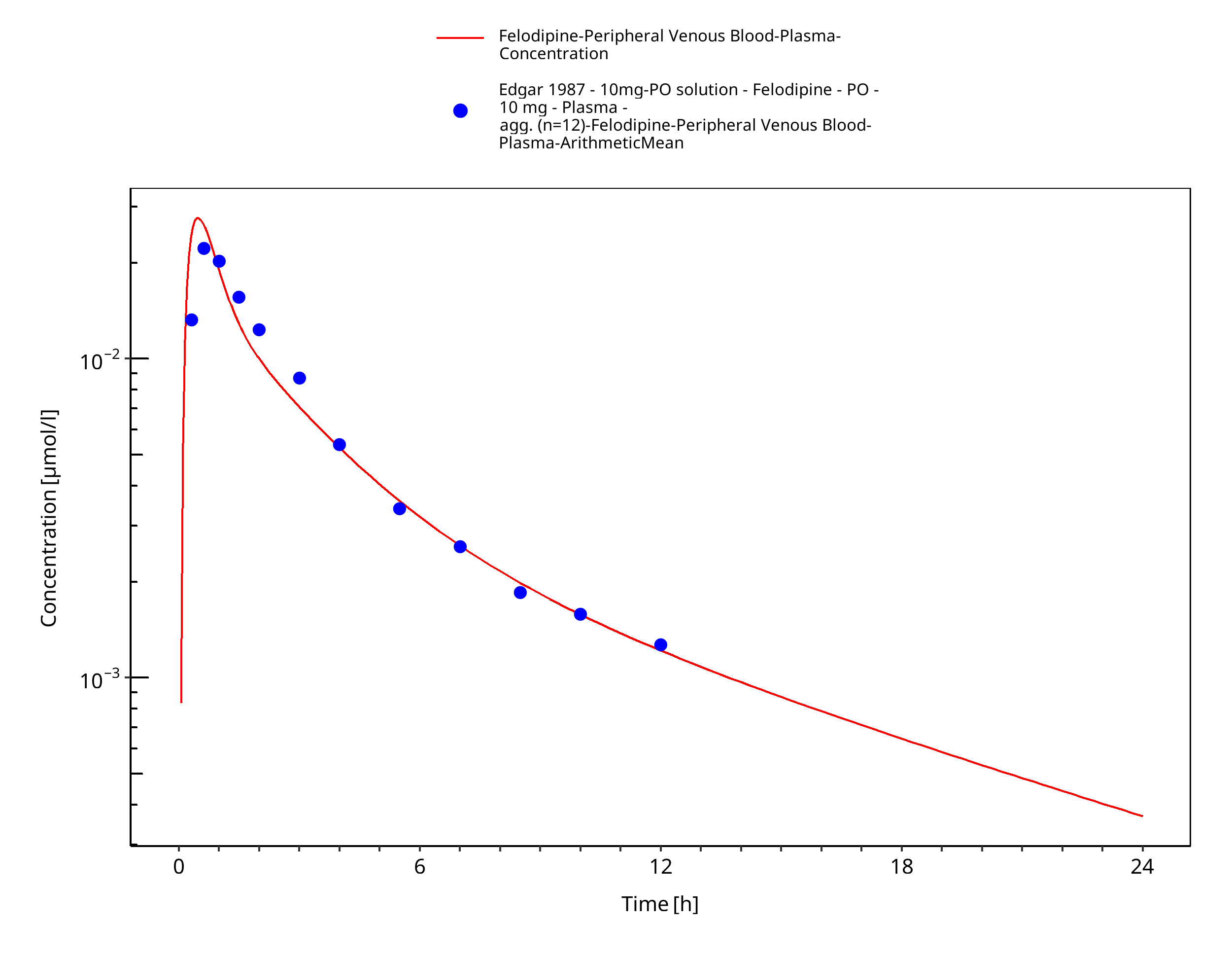
Figure 3-4: Time Profile Analysis
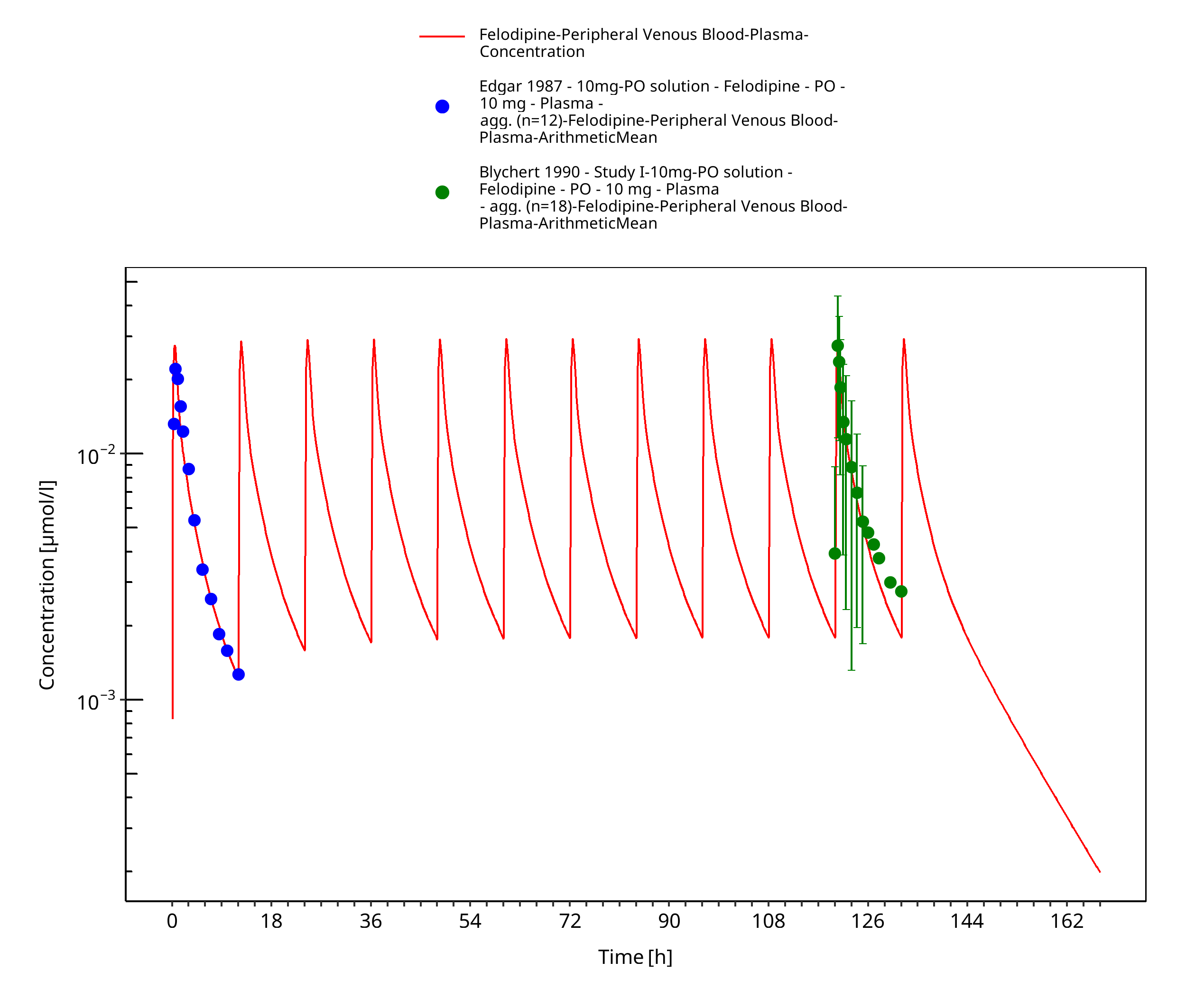
Figure 3-5: Time Profile Analysis
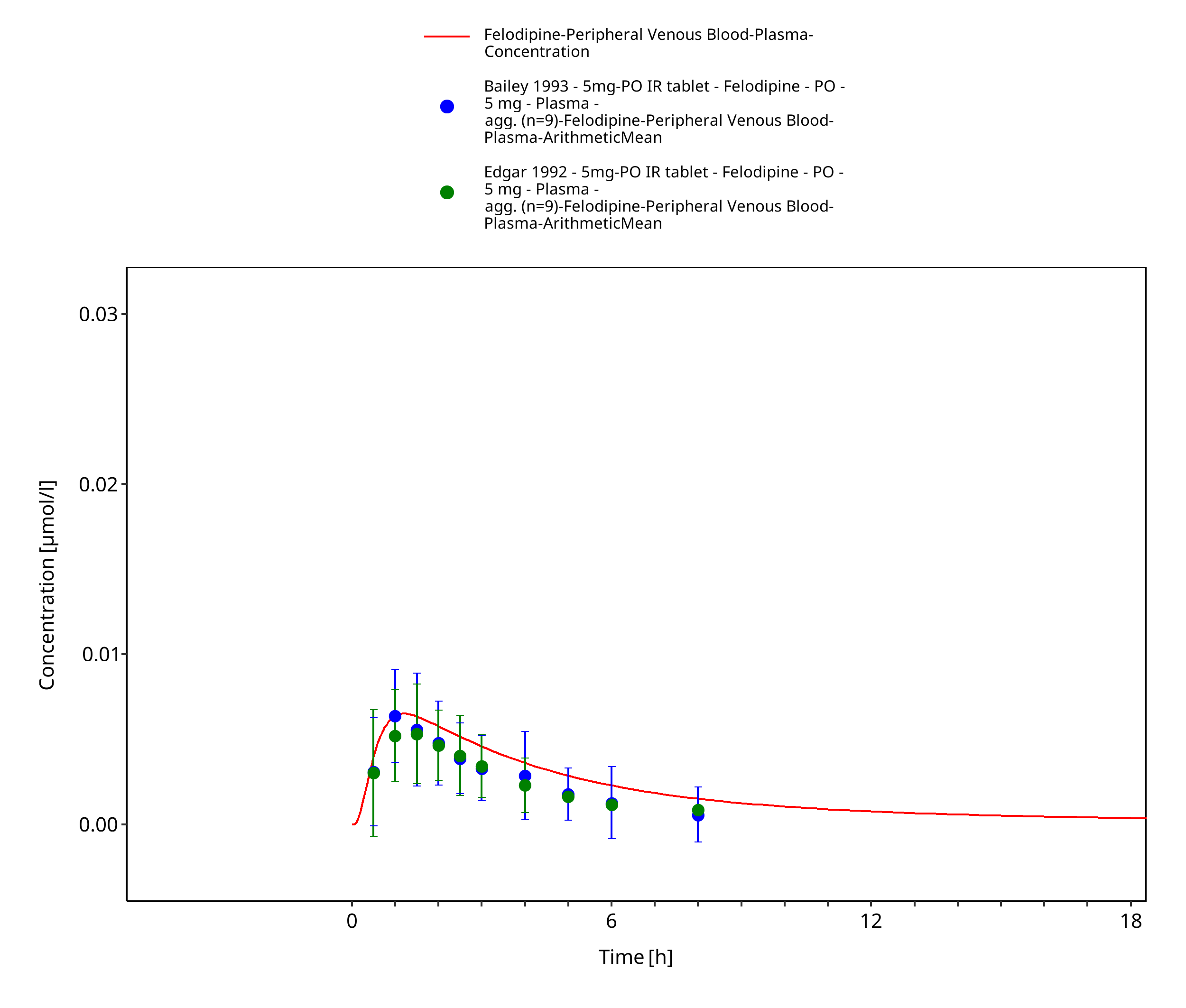
Figure 3-6: Time Profile Analysis
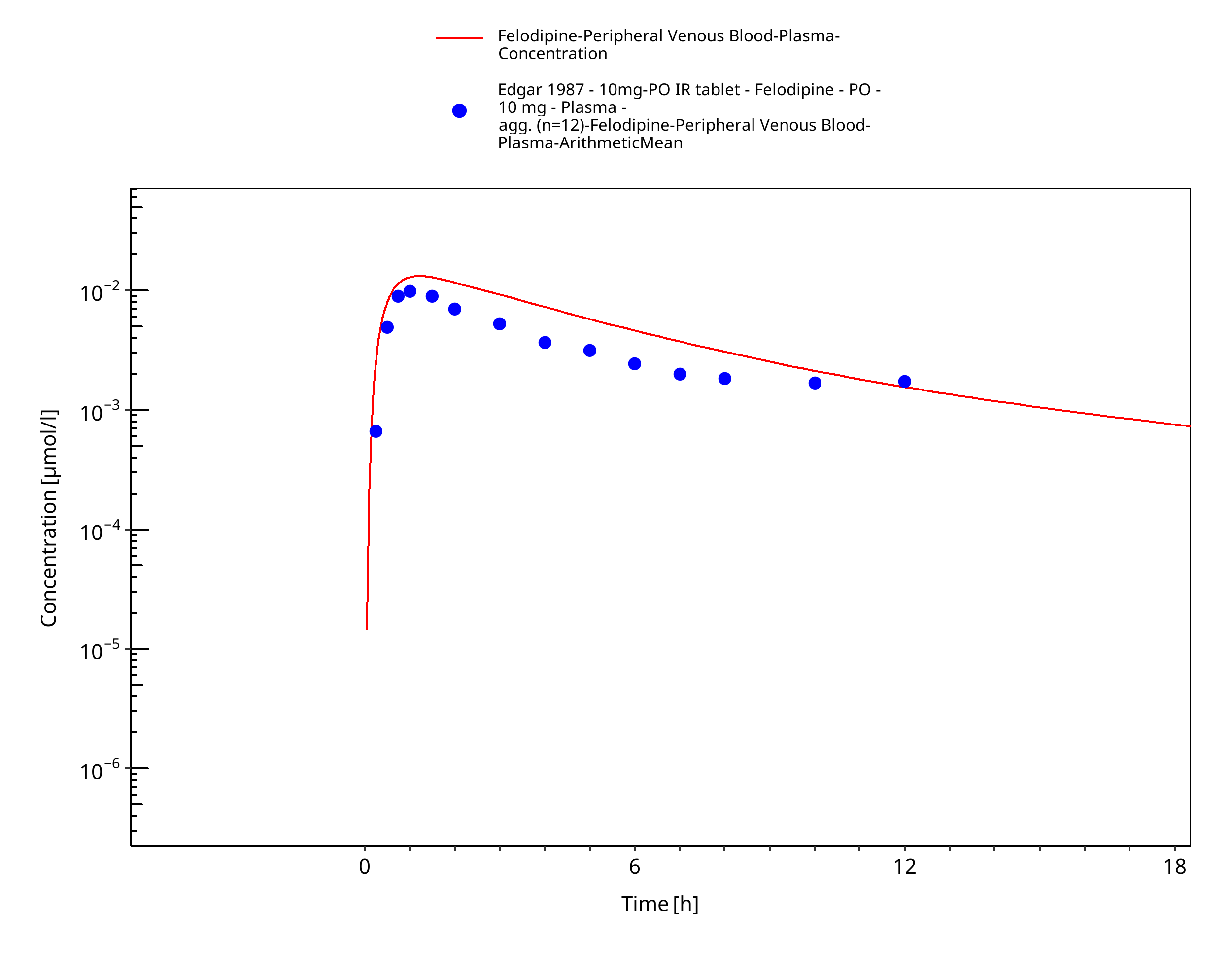
Figure 3-7: Time Profile Analysis
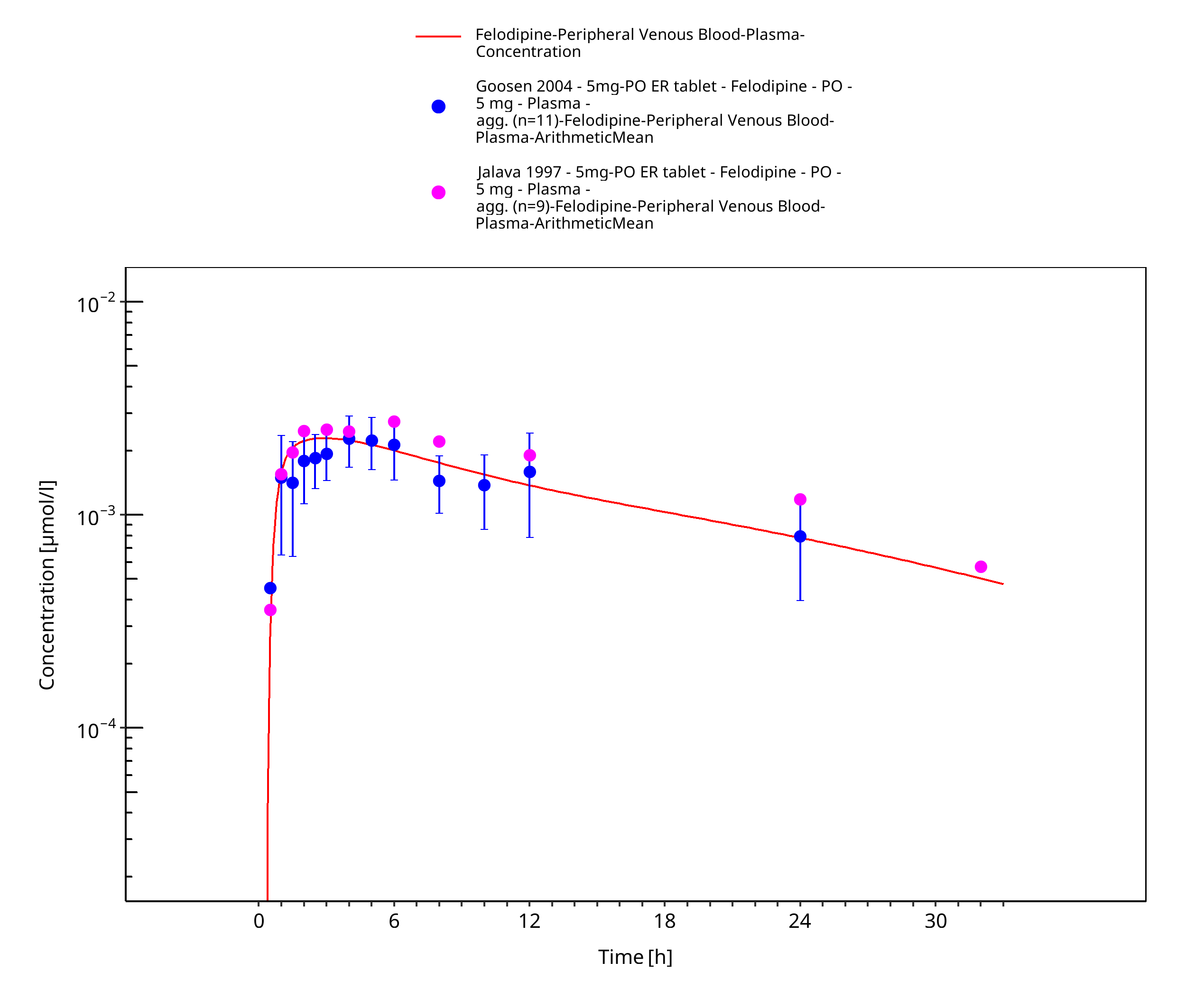
Figure 3-8: Time Profile Analysis
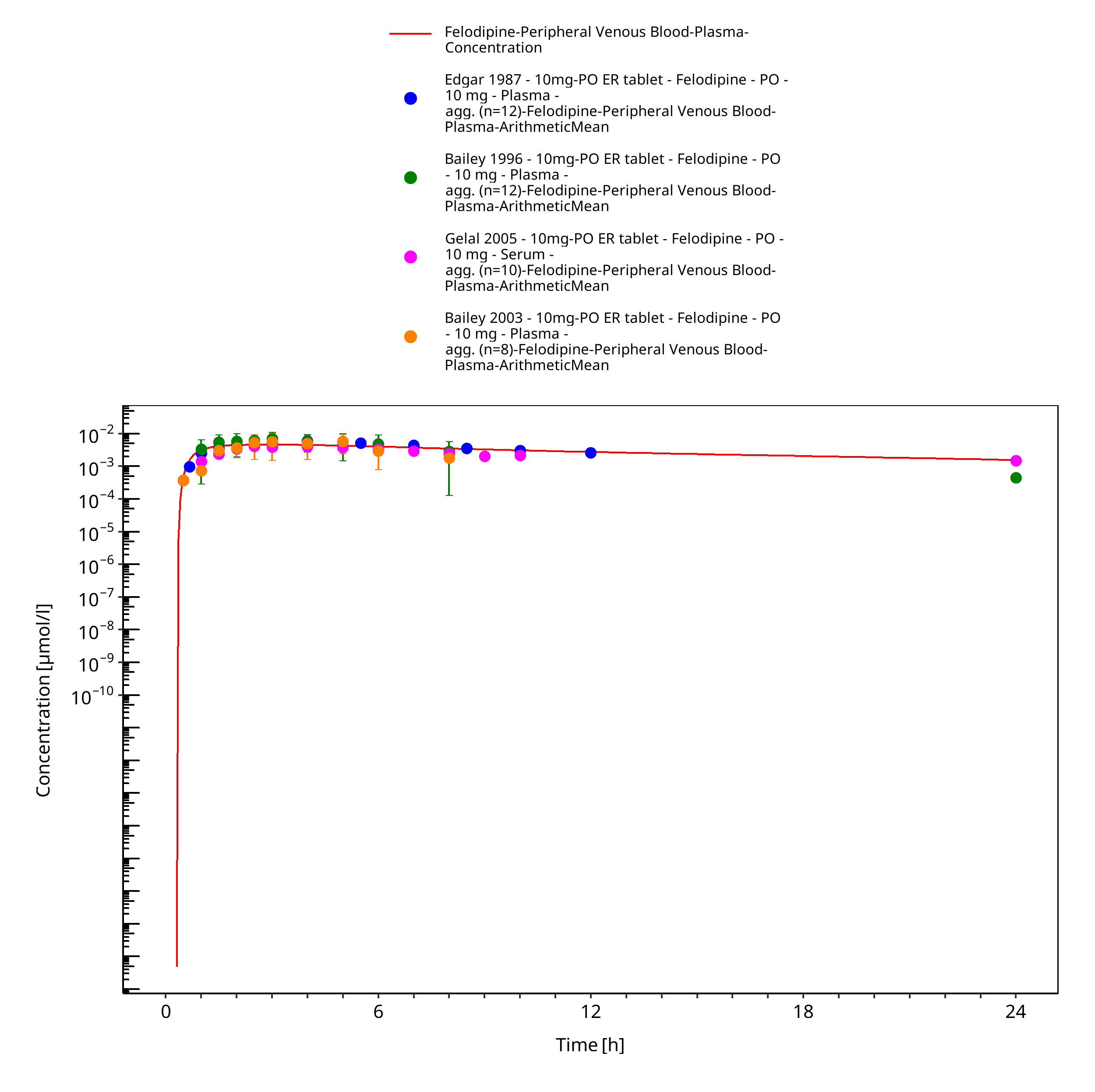
Figure 3-9: Time Profile Analysis
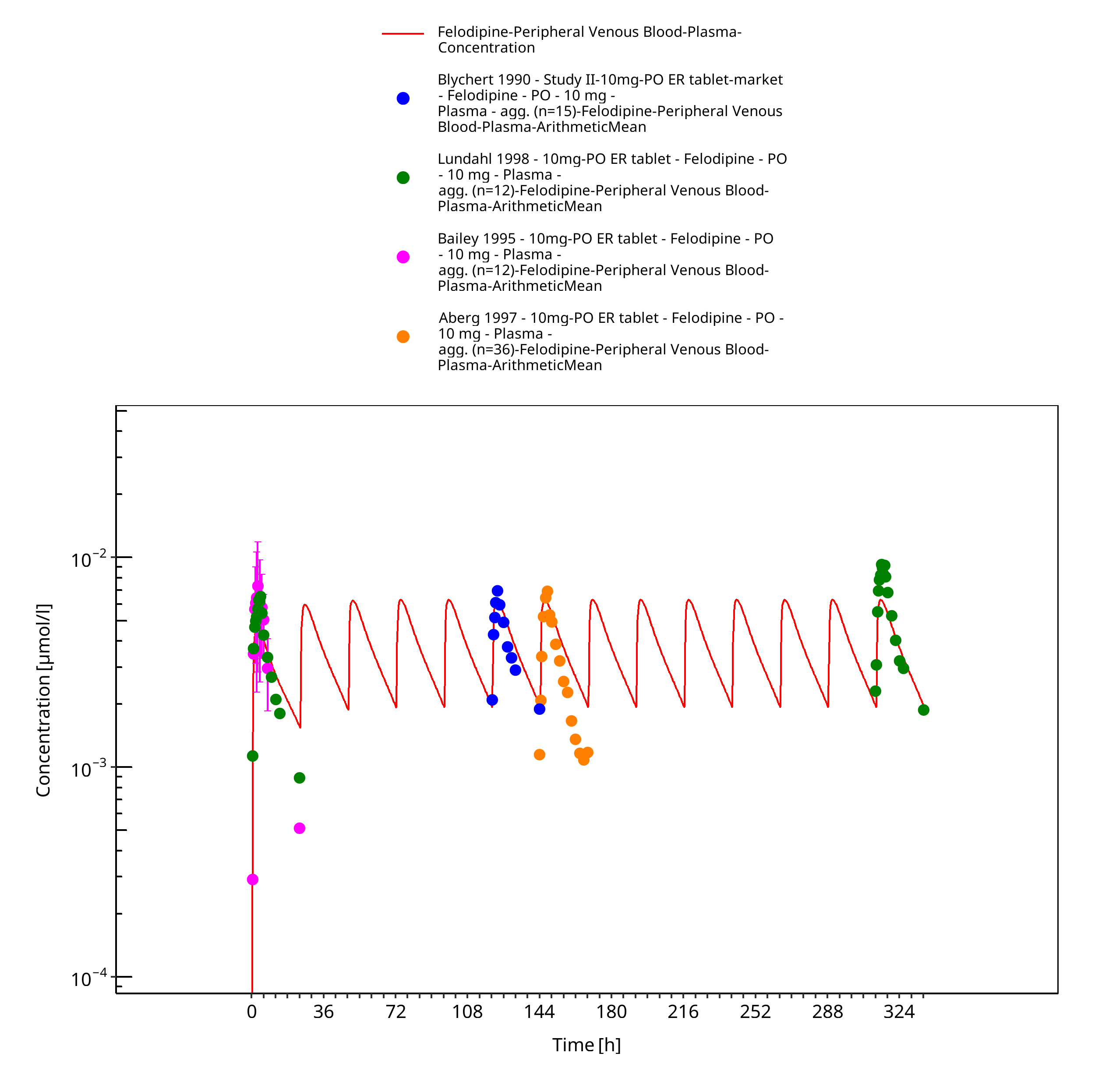
Figure 3-10: Time Profile Analysis
3.3.2 Model Verification¶
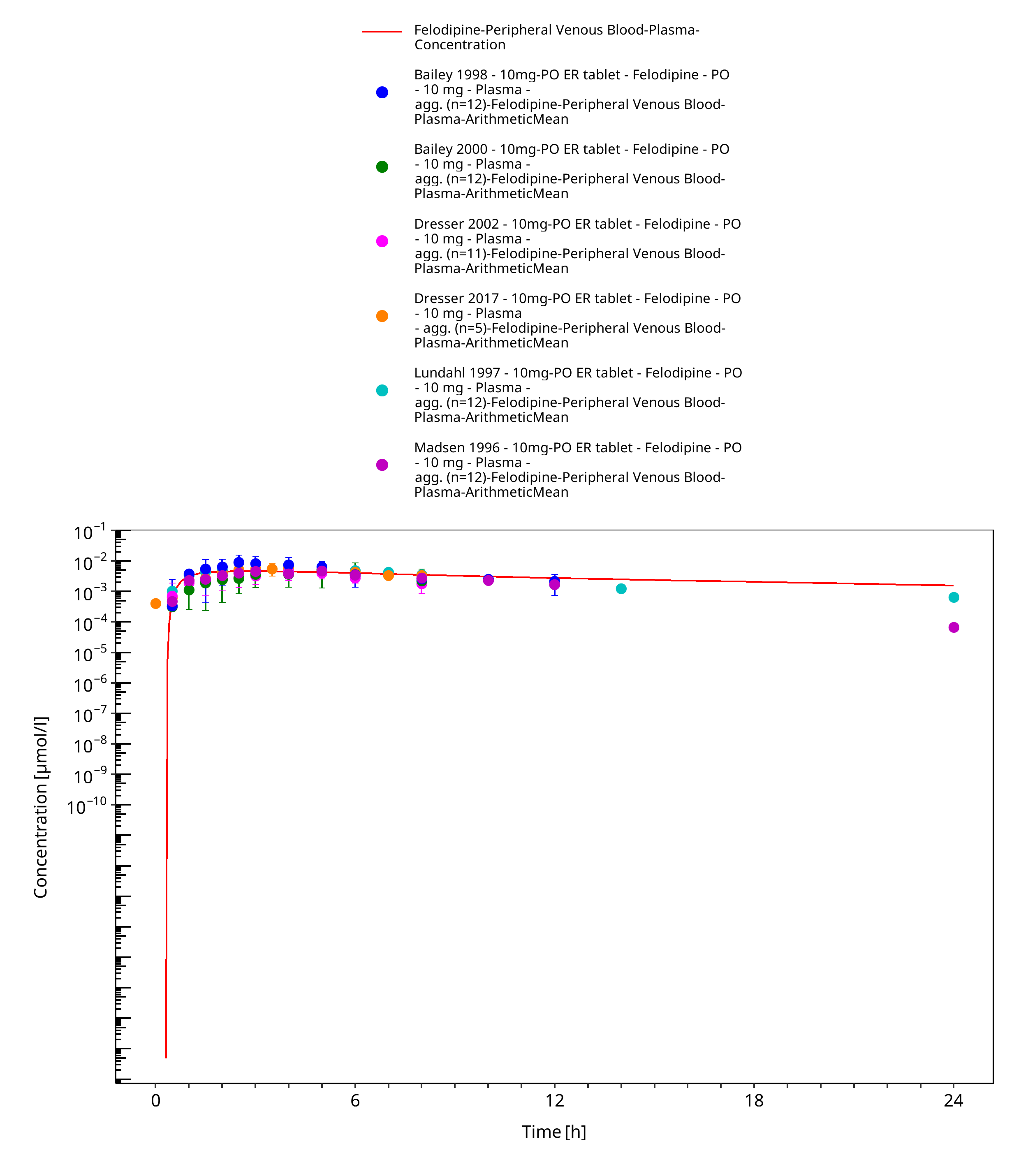
Figure 3-11: Time Profile Analysis
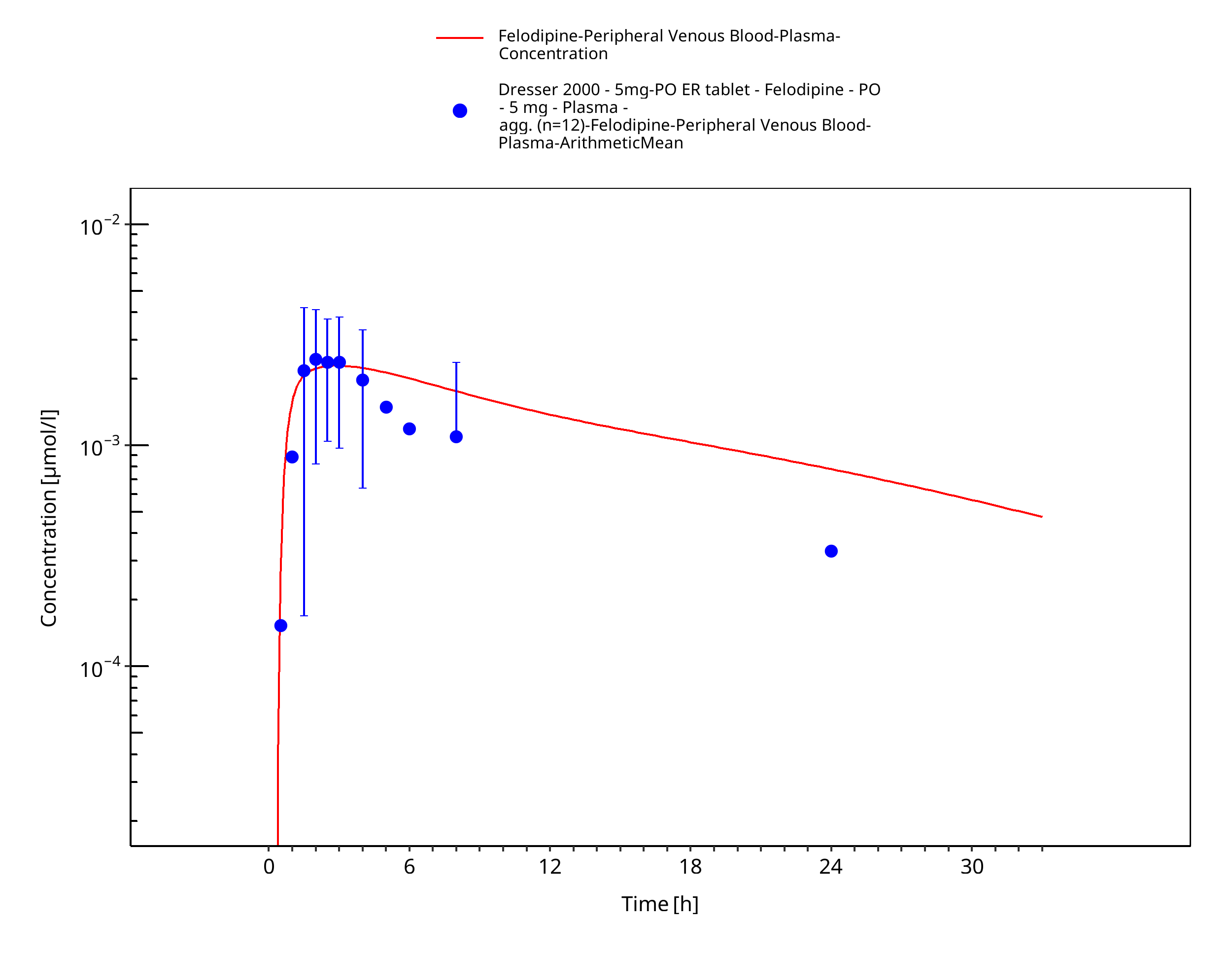
Figure 3-12: Time Profile Analysis
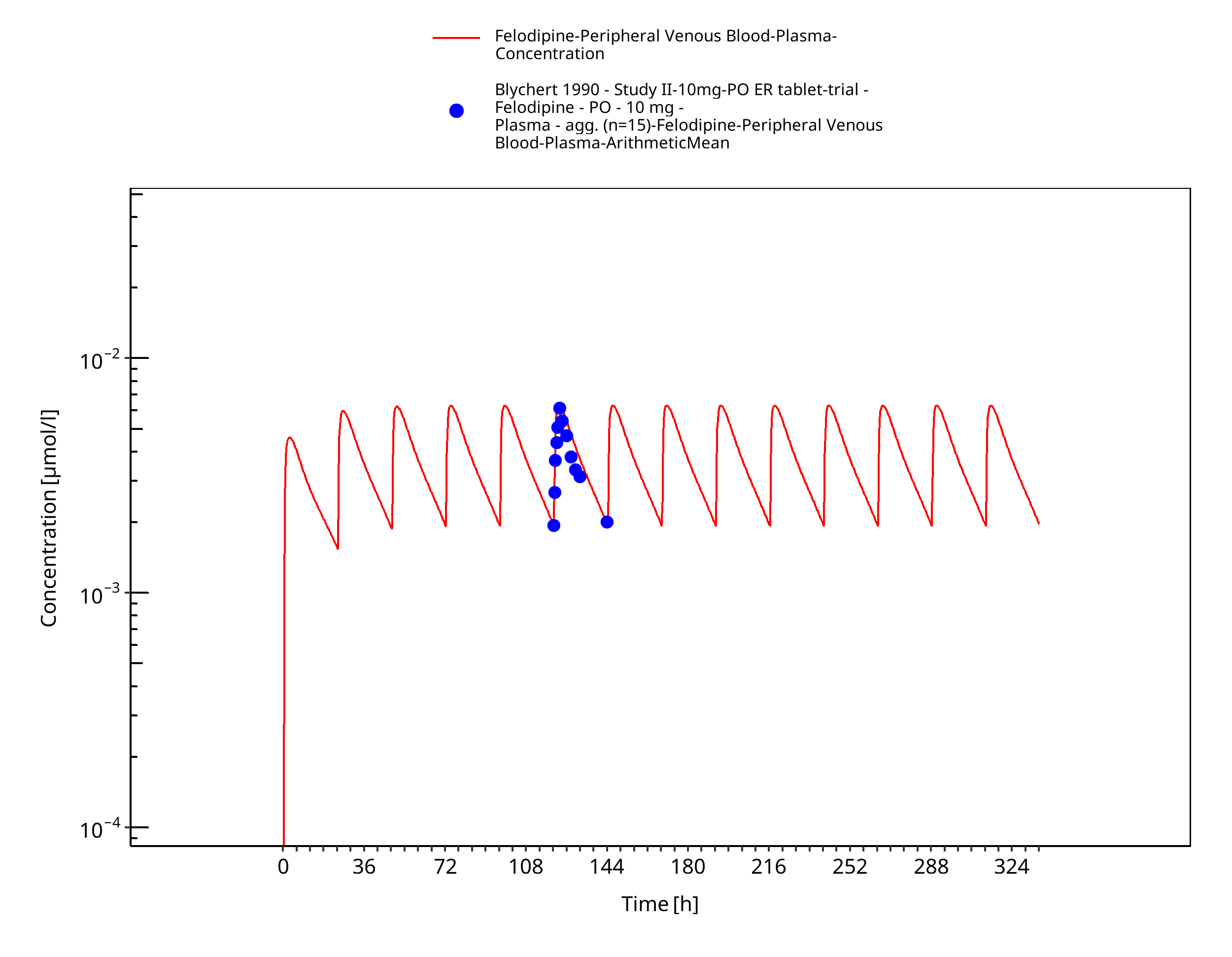
Figure 3-13: Time Profile Analysis
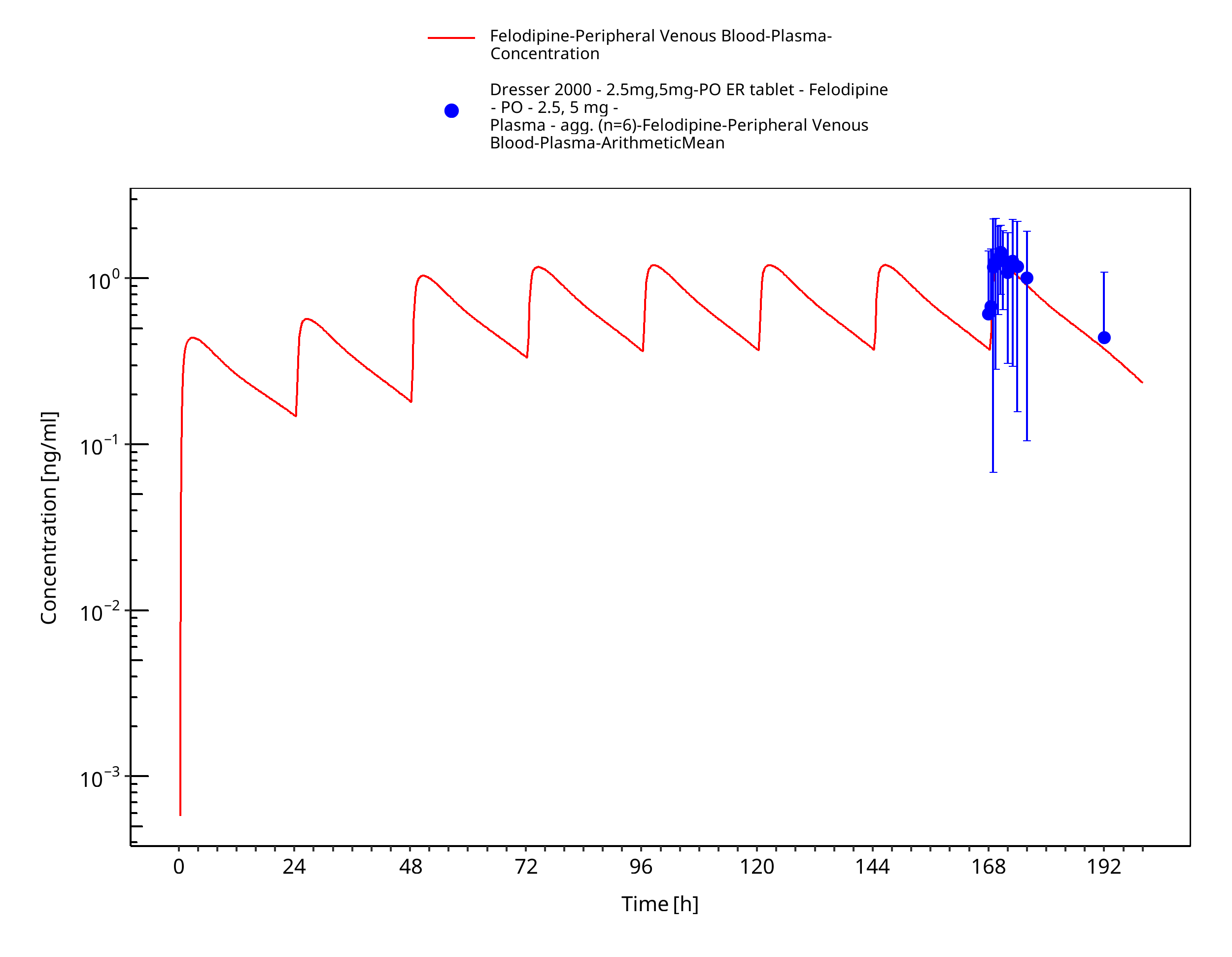
Figure 3-14: Time Profile Analysis
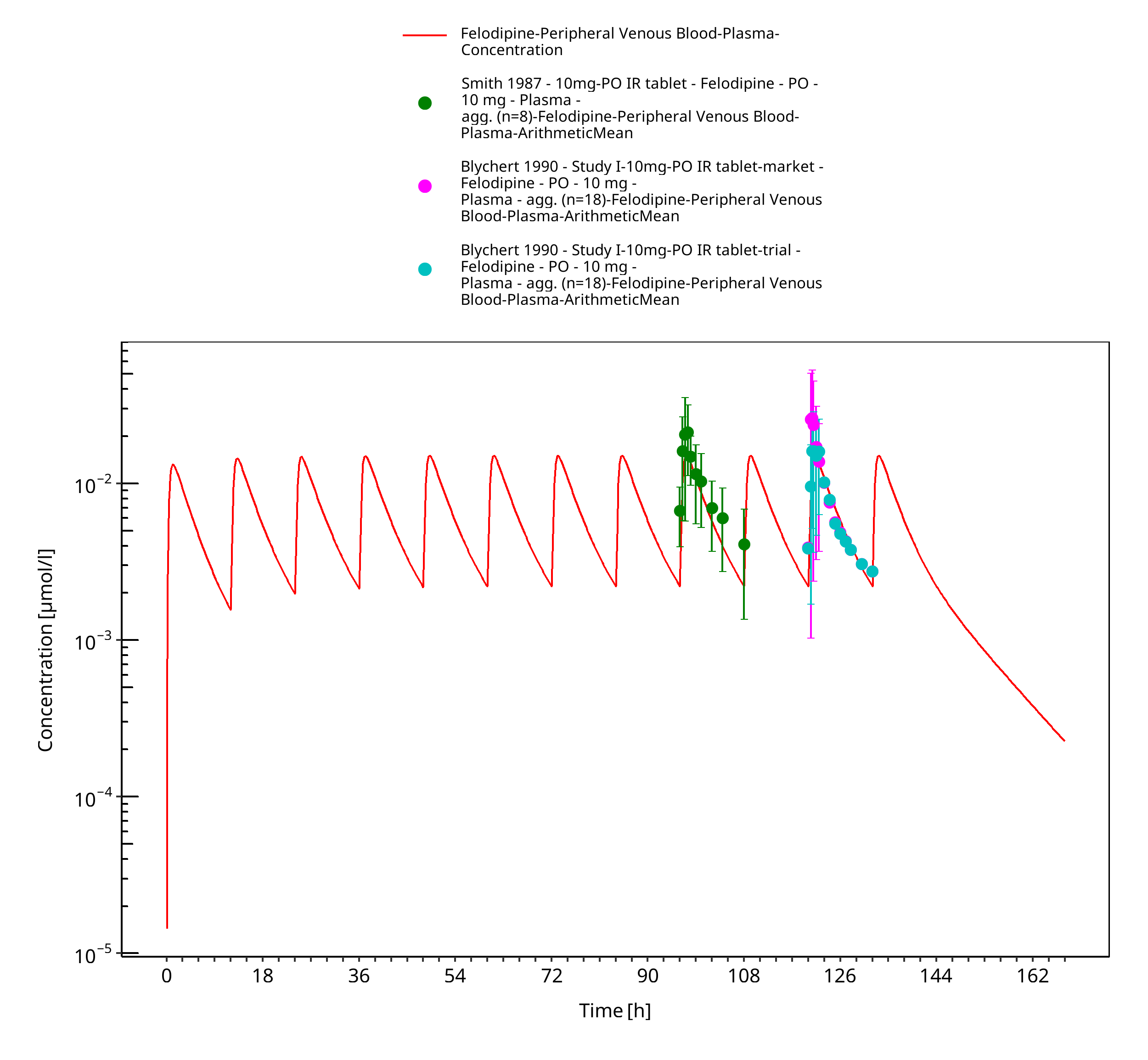
Figure 3-15: Time Profile Analysis
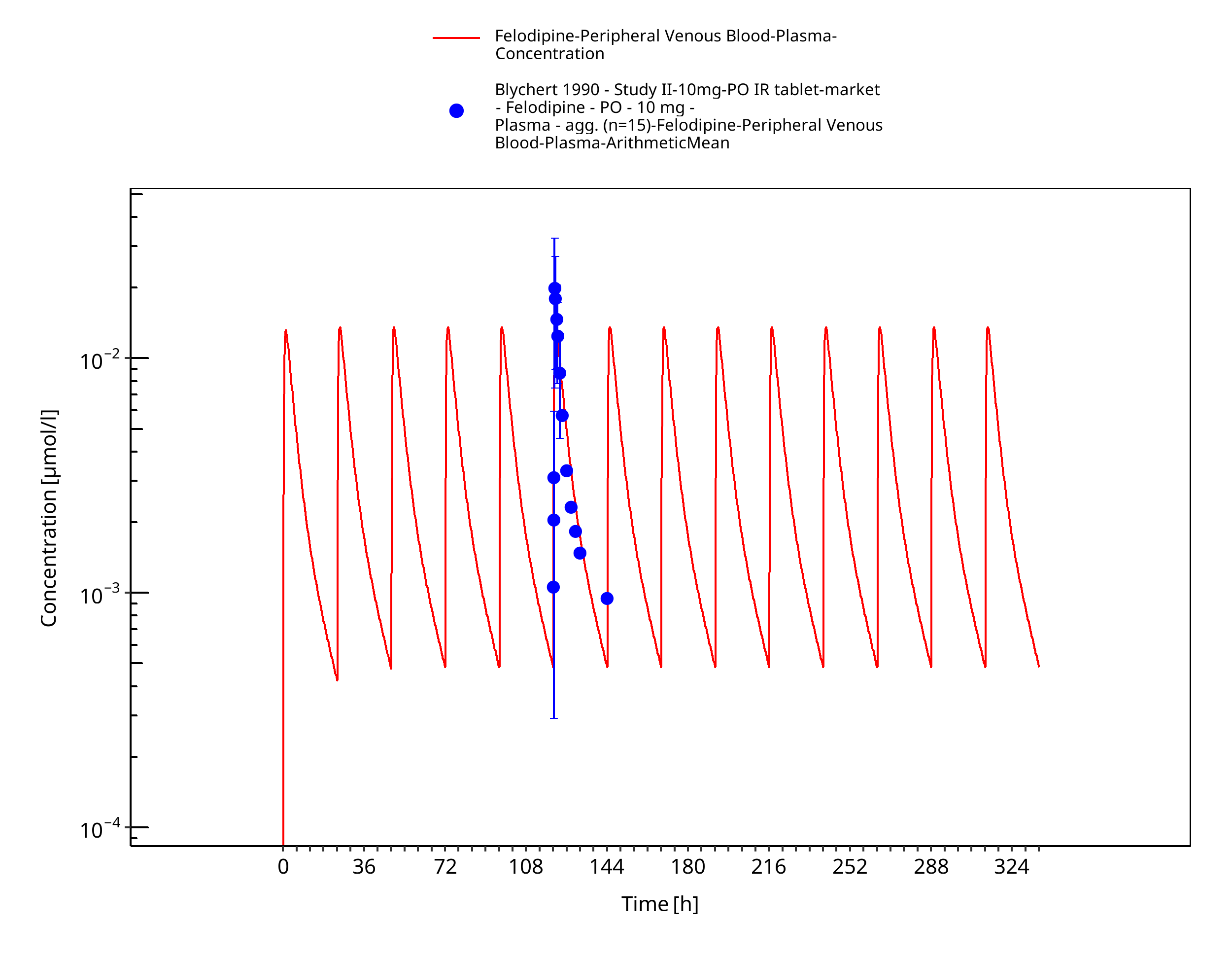
Figure 3-16: Time Profile Analysis
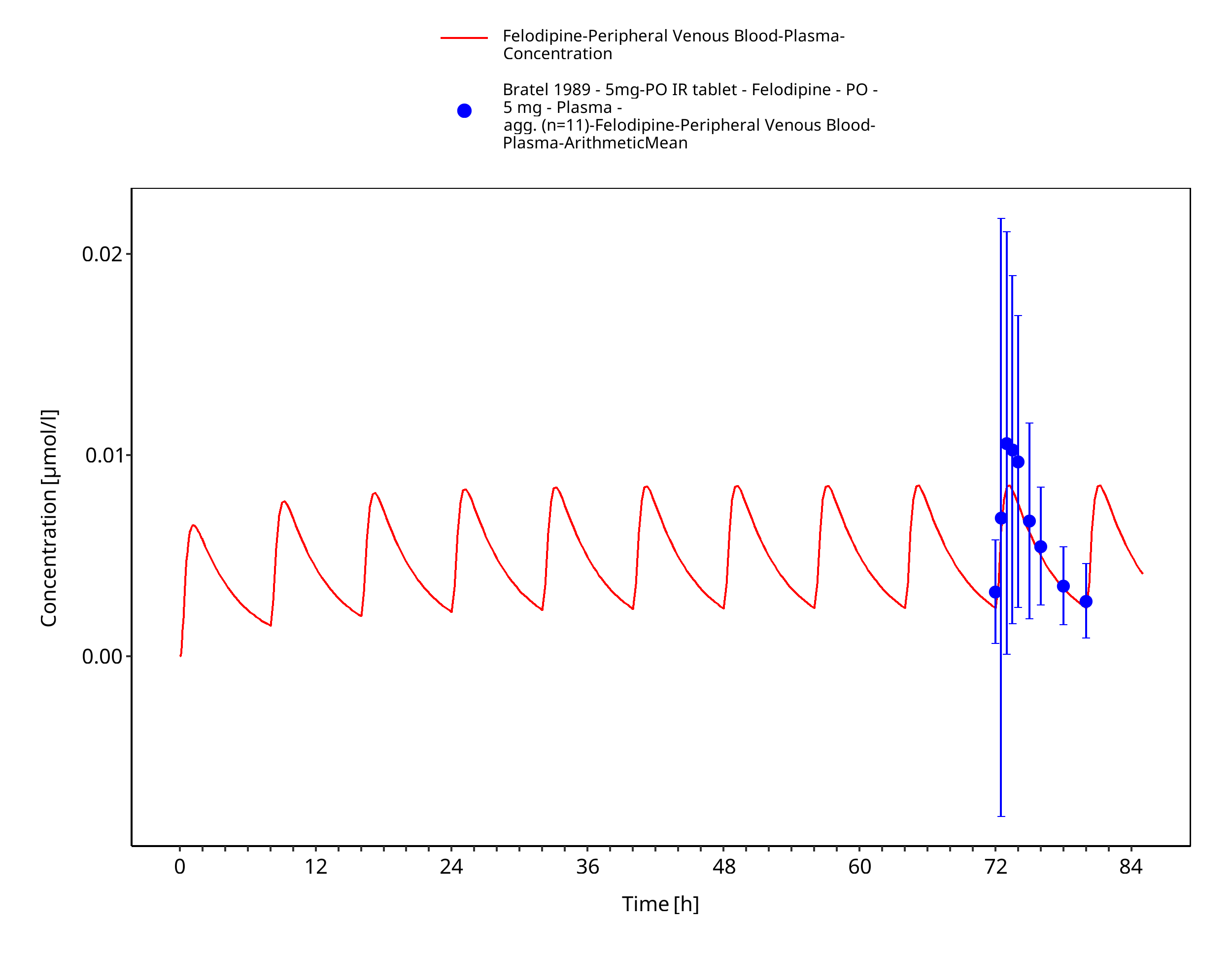
Figure 3-17: Time Profile Analysis
4 Conclusion¶
The herein presented PBPK model adequately describes the pharmacokinetics of felodipine in adults.
In particular, it applies quantitative metabolism by CYP3A4, and a second unknown hepatic enzyme. Thus, the model is fit for purpose to be applied for the investigation of drug-drug interactions with regard to its CYP3A4 metabolism.
5 References¶
Aberg 1997 Aberg J, Abrahamsson B, Grind M, Nyberg G, Olofsson B. Bioequivalence, pharmacokinetic and pharmacodynamic response to combined extended release formulations of felodipine and metoprolol in healthy volunteers. Eur J Clin Pharmacol. 1997;52(6):471-7.
Alskar 2018 Alskar LC, Keemink J, Johannesson J, Porter CJH, Bergstrom CAS. Impact of Drug Physicochemical Properties on Lipolysis-Triggered Drug Supersaturation and Precipitation from Lipid-Based Formulations. Mol Pharm. 2018;15(10):4733-44.
Bailey 1993 Bailey DG, Arnold JM, Munoz C, Spence JD. Grapefruit juice--felodipine interaction: mechanism, predictability, and effect of naringin. Clin Pharmacol Ther. 1993;53(6):637-42.
Bailey 1995 Bailey DG, Arnold JM, Bend JR, Tran LT, Spence JD. Grapefruit juice-felodipine interaction: reproducibility and characterization with the extended release drug formulation. Br J Clin Pharmacol. 1995;40(2):135-40.
Bailey 1996 Bailey DG, Bend JR, Arnold JM, Tran LT, Spence JD. Erythromycin-felodipine interaction: magnitude, mechanism, and comparison with grapefruit juice. Clin Pharmacol Ther. 1996;60(1):25-33.
Bailey 1998 Bailey DG, Kreeft JH, Munoz C, Freeman DJ, Bend JR. Grapefruit juice-felodipine interaction: effect of naringin and 6',7'-dihydroxybergamottin in humans. Clin Pharmacol Ther. 1998;64(3):248-56.
Bailey 2000 Bailey DG, Dresser GK, Kreeft JH, Munoz C, Freeman DJ, Bend JR. Grapefruit-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther. 2000;68(5):468-77.
Bailey 2003 Bailey D. Bergamottin, lime juice, and red wine as inhibitors of cytochrome P450 3a4 activity: comparison with grapefruit juice. Clinical Pharmacology & Therapeutics. 2003;73(6):529-37.
Blychert 1990 Blychert E, Wingstrand K, Edgar B, Lidman K. Plasma concentration profiles and antihypertensive effect of conventional and extended-release felodipine tablets. Br J Clin Pharmacol. 1990;29(1):39-45.
Bratel 1989 Bratel T, Billing B, Dahlqvist R. Felodipine reduces the absorption of theophylline in man. Eur J Clin Pharmacol. 1989;36(5):481-5.
Bu 2006 Bu HZ. A literature review of enzyme kinetic parameters for CYP3A4-mediated metabolic reactions of 113 drugs in human liver microsomes: structure-kinetics relationship assessment. Curr Drug Metab. 2006;7(3):231-49.
Dresser 2000 Dresser GK, Bailey DG, Carruthers SG. Grapefruit juice--felodipine interaction in the elderly. Clin Pharmacol Ther. 2000;68(1):28-34.
Dresser 2002 Dresser GK, Wacher V, Wong S, Wong HT, Bailey DG. Evaluation of peppermint oil and ascorbyl palmitate as inhibitors of cytochrome P4503A4 activity in vitro and in vivo. Clin Pharmacol Ther. 2002;72(3):247-55.
Dresser 2017 Dresser GK, Urquhart BL, Proniuk J, Tieu A, Freeman DJ, Arnold JM, et al. Coffee inhibition of CYP3A4 in vitro was not translated to a grapefruit-like pharmacokinetic interaction clinically. Pharmacol Res Perspect. 2017;5(5).
DrugBank DB01023 https://go.drugbank.com/drugs/DB01023
Edgar 1987 Edgar B, Lundborg P, Regårdh CG. Clinical pharmacokinetics of felodipine. A summary. Drugs. 1987;34 Suppl 3:16-27.
Edgar 1992 Edgar B, Bailey D, Bergstrand R, Johnsson G, Regårdh CG. Acute effects of drinking grapefruit juice on the pharmacokinetics and dynamics of felodipine--and its potential clinical relevance. Eur J Clin Pharmacol. 1992;42(3):313-7.
Gelal 2005 Gelal A, Balkan D, Ozzeybek D, Kaplan YC, Gurler S, Guven H, et al. Effect of menthol on the pharmacokinetics and pharmacodynamics of felodipine in healthy subjects. Eur J Clin Pharmacol. 2005;60(11):785-90.
Goosen 2004 Goosen TC, Cillie D, Bailey DG, Yu C, He K, Hollenberg PF, et al. Bergamottin contribution to the grapefruit juice-felodipine interaction and disposition in humans. Clin Pharmacol Ther. 2004;76(6):607-17.
Jalava 1997 Jalava KM, Olkkola KT, Neuvonen PJ. Itraconazole greatly increases plasma concentrations and effects of felodipine. Clin Pharmacol Ther. 1997;61(4):410-5.
Kuepfer 2016 Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, et al. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacometrics Syst Pharmacol. 2016;5(10):516-31.
Lundahl 1997 Lundahl J, Regårdh CG, Edgar B, Johnsson G. Effects of grapefruit juice ingestion--pharmacokinetics and haemodynamics of intravenously and orally administered felodipine in healthy men. Eur J Clin Pharmacol. 1997;52(2):139-45.
Lundahl 1998 Lundahl JU, Regårdh CG, Edgar B, Johnsson G. The interaction effect of grapefruit juice is maximal after the first glass. Eur J Clin Pharmacol. 1998;54(1):75-81.
Madsen 1996 Madsen JK, Jensen JD, Jensen LW, Pedersen EB. Pharmacokinetic interaction between cyclosporine and the dihydropyridine calcium antagonist felodipine. Eur J Clin Pharmacol. 1996;50(3):203-8.
McPherson 2020 McPherson S, Perrier J, Dunn C, Khadra I, Davidson S, Ainousah B, et al. Small scale design of experiment investigation of equilibrium solubility in simulated fasted and fed intestinal fluid. Eur J Pharm Biopharm. 2020;150:14-23.
Meyer 2012 Meyer M, Schneckener S, Ludewig B, Kuepfer L, Lippert J. Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab Dispos. 2012;40(5):892-901.
Nishimura 2003 Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi. 2003;123(5):369-75.
Pandey 2013 Pandey MM, Jaipal A, Kumar A, Malik R, Charde SY. Determination of pK(a) of felodipine using UV-Visible spectroscopy. Spectrochim Acta A Mol Biomol Spectrosc. 2013;115:887-90.
Persson 2005 Persson EM, Gustafsson AS, Carlsson AS, Nilsson RG, Knutson L, Forsell P, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm Res. 2005;22(12):2141-51.
PK-Sim Ontogeny Database Version 7.3 https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/38cf71b384cfc25cfa0ce4d2f3addfd32757e13b/PK-Sim%20Ontogeny%20Database%20Version%207.3.pdf
Rodrigues 2003 Rodrigues E, Vilarem MJ, Ribeiro V, Maurel P, Lechner MC. Two CCAAT/enhancer binding protein sites in the cytochrome P4503A1 locus. Potential role in the glucocorticoid response. Eur J Biochem. 2003;270(3):556-64.
Scholz 2002 Scholz A, Abrahamsson B, Diebold SM, Kostewicz E, Polentarutti BI, Ungell AL, et al. Influence of hydrodynamics and particle size on the absorption of felodipine in labradors. Pharm Res. 2002;19(1):42-6.
Smith 1987 Smith SR, Wilkins MR, Jack DB, Kendall MJ, Laugher S. Pharmacokinetic interactions between felodipine and metoprolol. Eur J Clin Pharmacol. 1987;31(5):575-8.
Söderlind 2010 Söderlind E, Karlsson E, Carlsson A, Kong R, Lenz A, Lindborg S, et al. Simulating fasted human intestinal fluids: understanding the roles of lecithin and bile acids. Mol Pharm. 2010;7(5):1498-507.
Soons 1993 Soons PA, Cohen AF, Breimer DD. Comparative effects of felodipine, nitrendipine and nifedipine in healthy subjects: concentration-effect relationships of racemic drugs and enantiomers. Eur J Clin Pharmacol. 1993;44(2):113-20.
Takano 2016Takano J, Maeda K, Bolger MB, Sugiyama Y. The Prediction of the Relative Importance of CYP3A/P-glycoprotein to the Nonlinear Intestinal Absorption of Drugs by Advanced Compartmental Absorption and Transit Model. Drug Metab Dispos. 2016;44(11):1808-18.
Ushimura 2010 Uchimura T, Kato M, Saito T, Kinoshita H. Prediction of human blood-to-plasma drug concentration ratio. Biopharm Drug Dispos. 2010;31(5-6):286-97.
Walsky 2004 Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32(6):647-60.
Willmann 2007 Willmann S, Höhn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401-31.
Related Models¶
Explore other Cardiovascular Agents models in this library:
- Cimetidine - PBPK model and evaluation report
- Dapagliflozin - PBPK model and evaluation report
- Digoxin - PBPK model and evaluation report
- Fluconazole - PBPK model and evaluation report
- Itraconazole - PBPK model and evaluation report