Building and evaluation of a PBPK Model for carbamazepine in healthy adults¶
| Version | 2.0-OSP12.3 |
|---|---|
| based on Model Snapshot and Evaluation Plan | https://github.com/Open-Systems-Pharmacology/Carbamazepine-Model/releases/tag/v2.0 |
| OSP Version | 12.3 |
| Qualification Framework Version | 3.7 |
This evaluation report and the corresponding PK-Sim project file are stored at:
https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library
Table of Contents¶
- 1 Introduction
- 2 Methods
- 2.1 Modeling Strategy
- 2.2 Data
- 2.3 Model Parameters and Assumptions
- 3 Results and Discussion
- 3.1 Final input parameters
- 3.2 Diagnostics Plots
- 3.3 Concentration-Time Profiles
- 4 Conclusion
- 5 References
1 Introduction¶
Carbamazepine, sold under the trade name Tegretol® among others, is an anticonvulsant medication used primarily to treat epilepsy and neuropathic pain. Other indications include schizophrenia where it is used as an adjunctive treatment along with other medications, and bipolar disorder where it is used as a second-line agent. Carbamazepine is typically taken by mouth on empty stomach or together with meals, depending on the administered formulation.
Carbamazepine is extensively metabolized by various enzymes including CYP2B6, 2C8, 3A4, and UGT2B7 (Kerr 1994, Pelkonen 2001, Staines 2004). Following oral administration the major dose fraction is metabolized to carbamazepine-10,11-epoxide (Eichelbaum 1985, Tomson 1983). This reaction is mainly catalyzed by CYP3A4, with some contribution from CYP2C8 (Kerr 1994). After oral administration, a minor fraction of the dose (approximately 1 - 3%) is excreted unchanged in urine (Bernus 1994, Morselli 1975), while approximately 1% of the dose can be recovered as unchanged drug in the bile (Terhaag 1978).
Carbamazepine is classified by the U.S. Food and Drug Administration (FDA) as a strong CYP3A4 and CYP2B6 inducer and hence induces its own metabolism.
The herein presented model was developed independently of the model reported by Fuhr et al. (Fuhr 2021). The main difference between the two models pertains to the metabolite carbamazepine-10,11-epoxide, which is included as separate compound in the model by Fuhr et al. (Fuhr 2021), but not modeled in the herein presented model. Another structural model differences concerns the enzymatic elimination pathways of carbamazepine; the model by Fuhr et al. (Fuhr 2021) includes five different metabolism pathways, whereas the herein presented model includes three different metabolism pathways. Additionally, the parameterization of CYP2B6 and 3A4 induction differs between the two models.
2 Methods¶
2.1 Modeling Strategy¶
The general workflow for building an adult PBPK model has been described by Kuepfer et al. (Kuepfer 2016). Relevant information on the anthropometry (height, weight) was gathered from the respective clinical study, if reported. Information on physiological parameters (e.g. blood flows, organ volumes, hematocrit) in adults was gathered from the literature and has been incorporated in PK-Sim®) as described previously (Willmann 2007). The applied activity and variability of plasma proteins and active processes that are integrated into PK-Sim® are described in the publicly available 'PK-Sim® Ontogeny Database Version 7.3' (PK-Sim Ontogeny Database Version 7.3).
The PBPK model was developed based on publicly available pharmacokinetic data of adult healthy subjects covering a carbamazepine dose range from 10 to 800 mg following intravenous administration or oral administration as liquid oral dosage form, immediate release (IR) tablet or extended release (XR) formulations in the fasted state. The carbamazepine PBPK model includes metabolism by CYP2B6, CYP3A4, and UGT2B7, unchanged renal excretion, and induction of CYP2B6 and 3A4 by carbamazepine. Pharmacokinetics of carbamazepine following administration in the fed state was not considered in the herein presented model. Furthermore, the metabolite carbamazepine-10,11-epoxide was not modeled as separate compound.
Unknown parameters (see below) were identified using the Parameter Identification module provided in PK-Sim®. Structural model selection was mainly guided by visual inspection of the resulting description of data and biological plausibility. Several parameter identifications were conducted to optimize unknown parameters. In a first step, lipophilicity and enzymatic clearances (catalyzed by CYP3A4, CYP2B6 and UGT2B7) were optimized using observed plasma concentration-time profile data following administration of carbamazepine intravenously or orally as syrup. In a second parameter identification, enzymatic clearances were refined and optimized together with the glomerular filtration rate fraction of carbamazepine and the dissolution kinetics of the IR tablet using observed plasma concentration-time profiles and the dose fraction excreted unchanged in urine after single dose administration of various doses as IR tablet. Subsequently, the EC50 value of CYP3A4 induction was optimized using observed plasma concentration-time profile data after multiple dose administration of carbamazepine. In a final parameter identification, the dissolution kinetics and carbamazepine solubility of XR formulations were optimized.
Details about input data (physicochemical, in vitro and clinical) can be found in Section 2.2.
Details about the structural model and its parameters can be found in Section 2.3.
2.2 Data¶
2.2.1 In vitro / physicochemical Data¶
A literature search was performed to collect available information on physicochemical properties of carbamazepine. The information is summarized in the table below.
| Parameter | Unit | Value | Source | Description |
|---|---|---|---|---|
| MW | g/mol | 236.27 | DrugBank DB00564 | Molecular weight |
| logP (calculated) | 1.54 | Austin 2002 | Partition coefficient between octanol and water | |
| logP (calculated) | 2.1 | DrugBank DB00564 | Partition coefficient between octanol and water | |
| logP (calculated) | 2.45 | Fenet 2012 | Partition coefficient between octanol and water | |
| logP (calculated) | 2.77 | DrugBank DB00564 | Partition coefficient between octanol and water | |
| Solubility (pH) | µg/mL | 336 (6.2) | Annaert 2010 | Solubility in human intestinal fluid |
| Solubility (pH) | µg/mL | 283 (7.0) | Söderlind 2010 | Solubility in human intestinal fluid |
| Solubility (pH) | µg/mL | 306 (6.9) | Clarysse 2011 | Solubility in fasted human intestinal fluid |
| fu | 0.25 | Pynnönen 1977 | Fraction unbound in plasma of healthy subjects | |
| fu | 0.243 ± 0.013 [0.225 - 0.258]a | Morselli 1975 | Fraction unbound in plasma of healthy male subjects | |
| fu | 0.239 | Di Salle 1974 | Fraction unbound in plasma of normal subjects | |
| fu | 0.237 ± 0.031b | Vinçon 1987 | Fraction unbound in plasma of epileptic patients | |
| fu | 0.182 ± 0.05 [0.103 - 0.297]a | Hooper 1975 | Fraction unbound in plasma of normal subjects | |
| Km CYP2B6 | µM | 420 | Pearce 2002 | CYP2B6 Michaelis-Menten constant |
| Vmax CYP2B6 | pmol/min/pmol rec enzyme | 0.429 | Pearce 2002 | in vitro metabolic rate constant for recombinant CYP2B6 |
| Km CYP2C8 | µM | 757 | Cazali 2003 | CYP2C8 Michaelis-Menten constant |
| Vmax CYP2C8 | pmol/min/pmol rec enzyme | 0.673 | Cazali 2003 | in vitro metabolic rate constant for recombinant CYP2C8 |
| Km CYP3A4c | µM | 282 | Pearce 2002 | CYP3A4 Michaelis-Menten constant |
| Km CYP3A4 (→CBZE)d | µM | 248 | Huang 2004 | CYP3A4 Michaelis-Menten constant |
| Km UGT2B7 | µM | 214 | Staines 2004 | UGT2B7 Michaelis-Menten constant |
| Vmax UGT2B7 | pmol/min/mg mic enzyme | 0.79 | Staines 2004 | in vitro metabolic rate constant for microsomal enzymes |
| Microsomal UGT2B7 | pmol/mg mic protein | 82.9 | Achour 2014 | Content of UGT2B7 proteins in liver microsomes |
| Intestinal permeability | cm/min | 0.0258 | Lennernäs 2007 | Transcellular intestinal permeability |
a denotes mean ± standard deviation [range]
b denotes mean ± standard deviation
c refers to CYP3A4-mediated reaction forming other metabolites than carbamazepine-10,11-epoxide
d refers to CYP3A4-mediated reaction forming carbamazepine-10,11-epoxide
2.2.2 Clinical Data¶
A literature search was conducted to collect available data on carbamazepine pharmacokinetics in healthy adult subjects after intravenous or oral administration in the fasted state.
The following studies were used for model building:
| Publication | Arm / Treatment / Information used for model building |
|---|---|
| Bernus 1994 | Healthy subjects receiving two oral doses of 600 mg carbamazepine as IR tablet (only pharmacokinetic data following the first dose were used for model building) |
| Gérardin 1976 | Healthy subjects receiving a single oral dose of 100 mg carbamazepine as IR tablet |
| Gérardin 1990 | Healthy subjects receiving a single oral dose of 100 mg [15N]-carbamazepine as suspension concomitantly with a single intravenous dose of 10 mg carbamazepine |
| McLean 2001 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine as XR formulation in fasted state |
| Møller 2001 | Healthy subjects receiving a multiple oral doses of carbamazepine, starting at 100 mg and escalating to 400 mg |
| Wada 1978 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine as syrup and IR tablet |
The following studies were used for model evaluation:
| Publication | Arm / Treatment / Information used for model building |
|---|---|
| Barzaghi 1987 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Bedada 2015 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Bedada 2016 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Bernus 1994 | Healthy subjects receiving two oral doses of 600 mg carbamazepine (only pharmacokinetic data following the second dose were used for model evaluation) |
| Bianchetti 1987 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Burstein 2000 | Healthy subjects receiving a multiple oral doses of carbamazepine, starting at 100 mg and escalating to 400 mg |
| Caraco 1995 | Healthy lean subjects receiving a single oral dose of 200 mg carbamazepine |
| Cawello 2000 | Healthy subjects receiving a multiple oral doses of carbamazepine, starting at 100 mg and escalating to 200 mg |
| Cotter 1977 | Healthy subject receiving a single oral dose of 800 mg carbamazepine |
| Dalton 1985a | Healthy subjects receiving a single oral dose of 600 mg carbamazepine |
| Dalton 1985b | Healthy subjects receiving a single oral dose of 600 mg carbamazepine |
| Eichelbaum 1985 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Elqidra 2004 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| European Patent Application EP 1044681 A2 | Healthy subjects receiving a single oral dose of 400 and 600 mg carbamazepine |
| Gérardin 1976 | Healthy subjects receiving a single oral dose of 200, and 600 mg carbamazepine |
| Ji 2008 | Healthy subjects receiving a multiple oral doses of carbamazepine, starting at 200 mg and escalating to 400 mg |
| Kayali 1994 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Kim 2005 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Kovacević 2009 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Levy 1975 | Healthy subjects receiving a single oral carbamazepine dose of 6 mg/kg body weight |
| Meyer 1996 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Meyer 1998 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Miles 1989 | Healthy subjects receiving a multiple oral doses of 300 and 400 mg carbamazepine |
| Morselli 1975 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Pynnönen 1977 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Rawlins 1975 | Healthy subject receiving a single oral dose of 50, 100, and 200 mg carbamazepine |
| Saint-Salvi 1987 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Stevens 1998 | Healthy subjects receiving multiple oral doses of 400 mg carbamazepine |
| Strandjord 1975 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
| Sumi 1987 | Healthy subjects receiving a single oral dose of 200 mg carbamazepine |
| Tomson 1983 | Healthy subject receiving a single oral doses of 200 mg carbamazepine |
| US Patent Application - US 2009/0169619 A1 | Healthy subjects receiving a single oral dose of 300 mg carbamazepine |
| Wong 1983 | Healthy subjects receiving a single oral dose of 400 mg carbamazepine |
2.3 Model Parameters and Assumptions¶
2.3.1 Absorption¶
Absorption of carbamazepine from the gastrointestinal tract can be fully explained by passive diffusion; active uptake by drug transporters does not seem to play a role. Intestinal permeability was observed to be not a rate-limiting step in drug absorption. The solubility of carbamazepine following administration of the IR tablet was fixed to the mean value (308 mg/L at a pH of 6.7) reported by several studies in fasted human intestinal fluid (Annaert 2010, Söderlind 2010, Clarysse 2011).
2.3.2 Distribution¶
Plasma protein binding of carbamazepine was fixed to 75.7% as reported by Morselli et al. for healthy subjects (Morselli 1975). The distribution of carbamazepine throughout the body was found to be best described by the partition coefficient calculation by Rodgers and Rowlands and cellular permeability calculation by PK-Sim Standard.
2.3.3 Metabolism, Excretion and Induction¶
Metabolism¶
Carbamazepine metabolism is complex involving multiple enzymes with more than 30 metabolites identified (Lertratanangkoon 1982). Several in vitro studies suggest involvement of CYP1A2, 2A6, 2B6, 2C8, 2E1, 3A4, and UGT2B7 in carbamazepine metabolism (Cazali 2003, Kerr 1994, Pearce 2002, Pelkonen 2001, Staines 2004).
In various in vitro assays, the biotransformation to the main metabolite, carbamazepine-10,11-epoxide, appears to be mainly catalyzed by CYP3A4 with minimal contribution by CYP2C8 (Cazali 2003, Egnell 2003, Kerr 1994). For example, Egnell et al. report that, at equimolar amounts of recombinantly expressed CYP enzymes, the activity of CYP3A4 towards carbamazepine was more than 20-fold higher than that of CYP2C8 (Egnell 2003). Therefore, carbamazepine epoxidation was modeled via CYP3A4 only.
Further oxidative metabolism pathways include 2- and 3-hydroxylation. The formation of 2-hydroxycarbamazepine is mediated by several CYP enzymes in vitro (including CYP1A2, 2A6, 2B6, 2E1, and 3A4); though, the contribution of any of these isoforms does not exceed 50% of the total formation (Pearce 2002). In experiments with liver slices, 2-hydroxylation appears to be a minor elimination pathway (1-2 % of total clearance) as reported by Pelkonen et al. (Pelkonen 2001). Hence, 2-hydroxylation was not accounted for in the PBPK model.
The formation of 3-hydroxycarbamazepine also appears to constitute a minor metabolism pathway (Pelkonen 2001); still, in human liver microsomes, 3-hydroxycarbamazepine was formed at rates ~25 times greater than those of 2-hydroxycarbamazepine (Pearce 2002). The responsible enzyme for 3-hydroxylation in vitro seems to be CYP2B6, although a minor contribution by CYP1A2, 2A6, and 3A4 cannot be ruled out (Pearce 2002). In the PBPK model, 3-hydroxylation was implemented as CYP2B6-mediated reaction.
N-glucuronidation of carbamazepine in human liver microsomes and baculovirus-infected insect cells expressing human UGTs was also observed with UGT2B7 appearing to be the responsible enzyme for this reaction (Staines 2004). Thus, the PBPK model also includes UGT2B7-mediated N-glucuronidation of carbamazepine.
In summary, the following three metabolic pathways, each mediated by a specific enzyme, were implemented in the PBPK model:
- 10,11-epoxidation via CYP3A4
- 3-hydroxylation via CYP2B6
- N-glucuronidation via UGT2B7
Since no clinical mass balance data were found for these three pathways, the following clearance kinetics in human liver microsomes reported for each pathway were initially implemented in the PBPK model:
| Biotransformation pathway | Km [µM] | Vmax [pmol/min/mg microsomal protein] | Source |
|---|---|---|---|
| 10,11-epoxidation | 808 | 726 | Sakamoto 2013 |
| 3-hydroxylation | 235 | 49.0 | Pearce 2002 |
| N-glucuronidation | 234 | 3.5 | Staines 2004 |
The following enzymatic content in human liver microsomes was assumed:
| Enzyme | Enzyme content [pmol/mg microsomal protein] | Source |
|---|---|---|
| CYP3A4 | 108 | Rodrigues 1999 |
| CYP2B6 | 39 | Rodrigues 1999 |
| UGT2B7 | 82.9 | Achour 2014 |
The expression profiles for these enzymes were loaded from the 'PK-Sim® Ontogeny Database Version 7.3' (PK-Sim Ontogeny Database Version 7.3) using RT-PCR as data source for each enzyme.
Upon implementation of these enzyme clearance pathways, it was seen that total clearance was slightly overestimated in the PBPK model. Therefore, the kcat values of each enzyme were optimized during parameter identification; to respect the initial mass balance of these biotransformation reactions as reported in human liver microsomes, the kcat values were not fitted independently but were varied together by the same factor.
Excretion¶
A minor fraction of the carbamazepine dose (approximately 1%) is excreted unchanged in urine (Bernus 1994, Morselli 1975). In the model, unchanged renal excretion was implemented as glomerular filtration with the parameter GFR fraction being fitted to the clinical excretion data reported by Bernus et al. (Bernus 1994).
Induction¶
Carbamazepine induces CYP2B6 and 3A4 via the CAR- and PXR-pathway (Faucette 2007, Williamson 2016). CYP2B6 induction was informed based on in vitro experiments conducted by Faucette et al. (Faucette 2004). These authors reported the induction of CYP2B6 activity at various carbamazepine concentrations in three preparations of primary human hepatocytes. The reported data suggest linear induction in the tested carbamazepine concentration range. A linear-mixed effects model was fitted to the reported data; the fitted slope was 0.149. To implement a linear induction in the PBPK model, the EC50 value of the Emax model was set to an arbitrarily high value (1000 µM) and Emax was then calculated as product of the fitted slope value and EC50 resulting in a value of 149.
CYP3A4 induction was initially parameterized based on internal in vitro experiments and calibrated with rifampicin induction data as described by Almond et al. (Almond 2016). This resulted in an EC50 of 63.0 µM and an Emax of 5.39. Simulated carbamazepine plasma concentrations in steady-state indicated that the induction was underestimated; therefore, the calibrated EC50 value was optimized during parameter identification, while the calibrated Emax value was kept fixed.
2.3.4 Automated Parameter Identification¶
The parameter identification tool in PK-Sim® has been used to estimate the model parameters described above. The result of the parameter identifications is shown in the table below:
| Model Parameter | Optimized Value | Unit |
|---|---|---|
Lipophilicity |
2.01 | |
kcat (CYP3A4) |
5.01 | 1/min |
kcat (CYP2B6) |
0.936 | 1/min |
kcat (UGT2B7) |
0.0669 | 1/min |
GFR fraction |
0.0240 | |
EC50 (CYP3A4) |
27.2 | µM |
Dissolution time (50% dissolved) (IR tablet, fasted) |
109 | min |
Dissolution shape (IR tablet, fasted) |
0.689 | |
Dissolution time (50% dissolved) (XR formulation, fasted) |
315 | min |
Dissolution shape (XR formulation, fasted) |
1.23 | |
Solubility at ref pH -- for XR formulations only |
546 | mg/L |
3 Results and Discussion¶
The PBPK model for carbamazepine was developed and evaluated using publicly available clinical pharmacokinetic data from studies listed in Section 2.2.2.
The next sections show:
- the final model parameters for the building blocks: Section 3.1.
- the overall goodness of fit: Section 3.2.
- simulated vs. observed concentration-time profiles for the clinical studies used for model building and for model verification: Section 3.3.
3.1 Final input parameters¶
The compound parameter values of the final PBPK model are illustrated below.
Compound: Carbamazepine¶
Parameters¶
| Name | Value | Value Origin | Alternative | Default |
|---|---|---|---|---|
| Solubility at reference pH | 308.3333 mg/l | Publication-Mean value of the following FaHIF solubility data reported in the literature: 336 µg/mL, pH 6.2 (Annaert 2010; DOI: 10.1016/j.ejps.2009.10.005); 283 µg/mL, pH 7.0 (Söderlind 2010; DOI: 10.1021/mp100144v); 306 mg/mL, pH 6.9 (Clarysse 2011; DOI: 10.1016/j.ejps.2011.04.016) | IR tablet (FaHIF) | True |
| Reference pH | 6.7 | Publication-Mean value of the following FaHIF solubility data reported in the literature: 336 µg/mL, pH 6.2 (Annaert 2010; DOI: 10.1016/j.ejps.2009.10.005); 283 µg/mL, pH 7.0 (Söderlind 2010; DOI: 10.1021/mp100144v); 306 mg/mL, pH 6.9 (Clarysse 2011; DOI: 10.1016/j.ejps.2011.04.016) | IR tablet (FaHIF) | True |
| Solubility at reference pH | 546.0199756643 mg/l | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-24 12:41 | XR tablet (fitted) | False |
| Reference pH | 6.7 | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-16 18:25 | XR tablet (fitted) | False |
| Lipophilicity | 2.0067753065 Log Units | Parameter Identification-Parameter Identification-Value updated from '001-5-3_CYP3A4_MM-kinetics_WithoutTablet' on 2022-02-21 16:49 | Optimized | True |
| Fraction unbound (plasma, reference value) | 0.243 | Publication-Morselli 1975 (DOI: 10.1007/978-3-642-85921-2_16) | Morselli 1975 | True |
| Is small molecule | Yes | |||
| Molecular weight | 236.2686 g/mol | Internet-DrugBank (https://go.drugbank.com/drugs/DB00564) | ||
| Plasma protein binding partner | Albumin |
Calculation methods¶
| Name | Value |
|---|---|
| Partition coefficients | Rodgers and Rowland |
| Cellular permeabilities | PK-Sim Standard |
Processes¶
Induction: CYP3A4-DMPK¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| EC50 | 27.193363407 µmol/l | Parameter Identification-Parameter Identification-Value updated from '003-1_from002-3-6_EC50' on 2022-02-24 10:23 |
| Emax | 5.3929777775 | Publication-In Vitro-DMPK measurement (internal data); the measured Emax was calibrated with rifampicin by using the Emax implemented in the rifampicin OSP model v1.2 according the the method described by Almond 2016 (DOI: 10.1124/dmd.115.066845) |
Systemic Process: Glomerular Filtration-Glomerular Filtration¶
Species: Human
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| GFR fraction | 0.0240108793 | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: UGT2B7-N-Glucuronidation_Staines2004¶
Molecule: UGT2B7
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 3.5 pmol/min/mg mic. protein | Publication-In Vitro-Staines 2004 (DOI: 10.1124/jpet.104.073114) |
| Content of CYP proteins in liver microsomes | 82.9 pmol/mg mic. protein | Publication-In Vitro-Achour 2014 (DOI: 10.1124/dmd.113.055632) |
| Km | 234 µmol/l | Publication-In Vitro-Staines 2004 (DOI: 10.1124/jpet.104.073114) |
| kcat | 0.0668699322 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: CYP2B6-3-Hydroxylation_Pearce2002¶
Molecule: CYP2B6
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 49 pmol/min/mg mic. protein | Publication-In Vitro-Pearce 2002 (DOI: 10.1124/dmd.30.11.1170) |
| Content of CYP proteins in liver microsomes | 39 pmol/mg mic. protein | Publication-In Vitro-Rodrigues 1999 (DOI: 10.1016/s0006-2952(98)00268-8) |
| Km | 235 µmol/l | Publication-Pearce 2002 (DOI: 10.1124/dmd.30.11.1170) |
| kcat | 0.9361790504 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: CYP3A4-Epoxidation_Sakamoto2013¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 726 pmol/min/mg mic. protein | Publication-In Vitro-Sakamoto 2013 (DOI: 10.1248/bpb.b13-00569) |
| Km | 808 µmol/l | Publication-In Vitro-Sakamoto 2013 (DOI: 10.1248/bpb.b13-00569) |
| kcat | 5.0088763476 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Induction: CYP2B6-Faucette2004¶
Molecule: CYP2B6
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| EC50 | 1000 µmol/l | Publication-Set to an arbitrarily high value to enable linear induction as suggested by Faucette 2004 (DOI: 10.1124/dmd.32.3.348); see evaluation report for details |
| Emax | 148.7284 | Publication-Linear-mixed effects model fitted to reported data by Faucette 2004 (DOI: 10.1124/dmd.32.3.348); see evaluation report for details |
Compound: [15N]-Carbamazepine¶
Parameters¶
| Name | Value | Value Origin | Alternative | Default |
|---|---|---|---|---|
| Solubility at reference pH | 308.3333 mg/l | Publication-Mean value of the following FaHIF solubility data reported in the literature: 336 µg/mL, pH 6.2 (Annaert 2010; DOI: 10.1016/j.ejps.2009.10.005); 283 µg/mL, pH 7.0 (Söderlind 2010; DOI: 10.1021/mp100144v); 306 mg/mL, pH 6.9 (Clarysse 2011; DOI: 10.1016/j.ejps.2011.04.016) | IR tablet (FaHIF) | True |
| Reference pH | 6.7 | Publication-Mean value of the following FaHIF solubility data reported in the literature: 336 µg/mL, pH 6.2 (Annaert 2010; DOI: 10.1016/j.ejps.2009.10.005); 283 µg/mL, pH 7.0 (Söderlind 2010; DOI: 10.1021/mp100144v); 306 mg/mL, pH 6.9 (Clarysse 2011; DOI: 10.1016/j.ejps.2011.04.016) | IR tablet (FaHIF) | True |
| Solubility at reference pH | 546.0199756643 mg/l | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-24 12:41 | XR tablet (fitted) | False |
| Reference pH | 6.7 | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-16 18:25 | XR tablet (fitted) | False |
| Lipophilicity | 2.0067753065 Log Units | Parameter Identification-Parameter Identification-Value updated from '001-5-3_CYP3A4_MM-kinetics_WithoutTablet' on 2022-02-21 16:49 | Optimized | True |
| Fraction unbound (plasma, reference value) | 0.243 | Publication-Morselli 1975 (DOI: 10.1007/978-3-642-85921-2_16) | Morselli 1975 | True |
| Is small molecule | Yes | |||
| Molecular weight | 236.2686 g/mol | Internet-DrugBank (https://go.drugbank.com/drugs/DB00564) | ||
| Plasma protein binding partner | Albumin |
Calculation methods¶
| Name | Value |
|---|---|
| Partition coefficients | Rodgers and Rowland |
| Cellular permeabilities | PK-Sim Standard |
Processes¶
Induction: CYP3A4-DMPK¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| EC50 | 27.193363407 µmol/l | Parameter Identification-Parameter Identification-Value updated from '003-1_from002-3-6_EC50' on 2022-02-24 10:23 |
| Emax | 5.3929777775 | Publication-In Vitro-DMPK measurement (internal data); the measured Emax was calibrated with rifampicin by using the Emax implemented in the rifampicin OSP model v1.2 according the the method described by Almond 2016 (DOI: 10.1124/dmd.115.066845) |
Systemic Process: Glomerular Filtration-Glomerular Filtration¶
Species: Human
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| GFR fraction | 0.0240108793 | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: UGT2B7-N-Glucuronidation_Staines2004¶
Molecule: UGT2B7
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 3.5 pmol/min/mg mic. protein | Publication-In Vitro-Staines 2004 (DOI: 10.1124/jpet.104.073114) |
| Content of CYP proteins in liver microsomes | 82.9 pmol/mg mic. protein | Publication-In Vitro-Achour 2014 (DOI: 10.1124/dmd.113.055632) |
| Km | 234 µmol/l | Publication-In Vitro-Staines 2004 (DOI: 10.1124/jpet.104.073114) |
| kcat | 0.0668699322 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: CYP2B6-3-Hydroxylation_Pearce2002¶
Molecule: CYP2B6
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 49 pmol/min/mg mic. protein | Publication-In Vitro-Pearce 2002 (DOI: 10.1124/dmd.30.11.1170) |
| Content of CYP proteins in liver microsomes | 39 pmol/mg mic. protein | Publication-In Vitro-Rodrigues 1999 (DOI: 10.1016/s0006-2952(98)00268-8) |
| Km | 235 µmol/l | Publication-Pearce 2002 (DOI: 10.1124/dmd.30.11.1170) |
| kcat | 0.9361790504 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Metabolizing Enzyme: CYP3A4-Epoxidation_Sakamoto2013¶
Molecule: CYP3A4
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| In vitro Vmax for liver microsomes | 726 pmol/min/mg mic. protein | Publication-In Vitro-Sakamoto 2013 (DOI: 10.1248/bpb.b13-00569) |
| Km | 808 µmol/l | Publication-In Vitro-Sakamoto 2013 (DOI: 10.1248/bpb.b13-00569) |
| kcat | 5.0088763476 1/min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
Induction: CYP2B6-Faucette2004¶
Molecule: CYP2B6
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| EC50 | 1000 µmol/l | Publication-Set to an arbitrarily high value to enable linear induction as suggested by Faucette 2004 (DOI: 10.1124/dmd.32.3.348); see evaluation report for details |
| Emax | 148.7284 | Publication-Linear-mixed effects model fitted to reported data by Faucette 2004 (DOI: 10.1124/dmd.32.3.348); see evaluation report for details |
Formulation: CBZ_tabletIR_fasted (Tegretol)¶
Type: Weibull
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Dissolution time (50% dissolved) | 109.3089775422 min | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
| Lag time | 0 min | |
| Dissolution shape | 0.6890123758 | Parameter Identification-Parameter Identification-Value updated from '002-3-6_from001-5-3_IRtablet-sd_Pint-FIX_FINAL' on 2022-02-23 17:18 |
| Use as suspension | Yes |
Formulation: CBZ_capsuleXR_fasted (Carbatrol)¶
Type: Weibull
Parameters¶
| Name | Value | Value Origin |
|---|---|---|
| Dissolution time (50% dissolved) | 315.2431776804 min | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-24 12:41 |
| Lag time | 0 min | |
| Dissolution shape | 1.2290186648 | Parameter Identification-Parameter Identification-Value updated from '004-2_from-003-1_XRtablet_fasted_solubility_FINAL' on 2022-03-24 12:41 |
| Use as suspension | Yes |
Formulation: Solution¶
Type: Dissolved
3.2 Diagnostics Plots¶
Below you find the goodness-of-fit visual diagnostic plots for the PBPK model performance of all data used presented in Section 2.2.2.
The first plot shows simulated versus observed plasma concentration, the second weighted residuals versus time.
Table 3-1: GMFE for Goodness of fit plot for concentration in plasma
| Group | GMFE |
|---|---|
| Carbamazepine, IV administration | 1.28 |
| Carbamazepine, PO administration as extended release formulation | 1.41 |
| Carbamazepine, PO administration as immediate release tablet | 1.40 |
| Carbamazepine, PO administration as liquid oral dosage form | 2.35 |
| All | 1.49 |
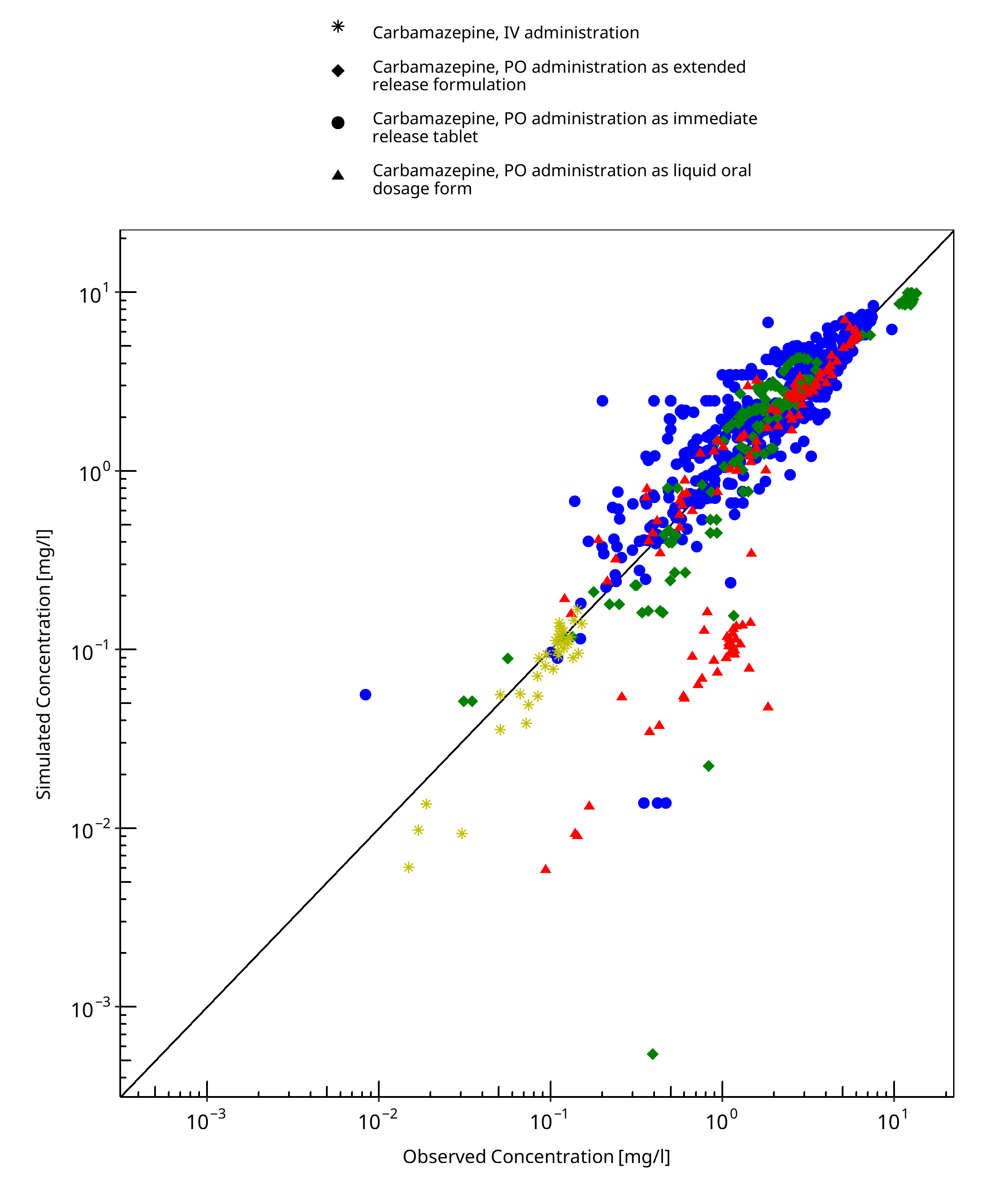
Figure 3-1: Goodness of fit plot for concentration in plasma
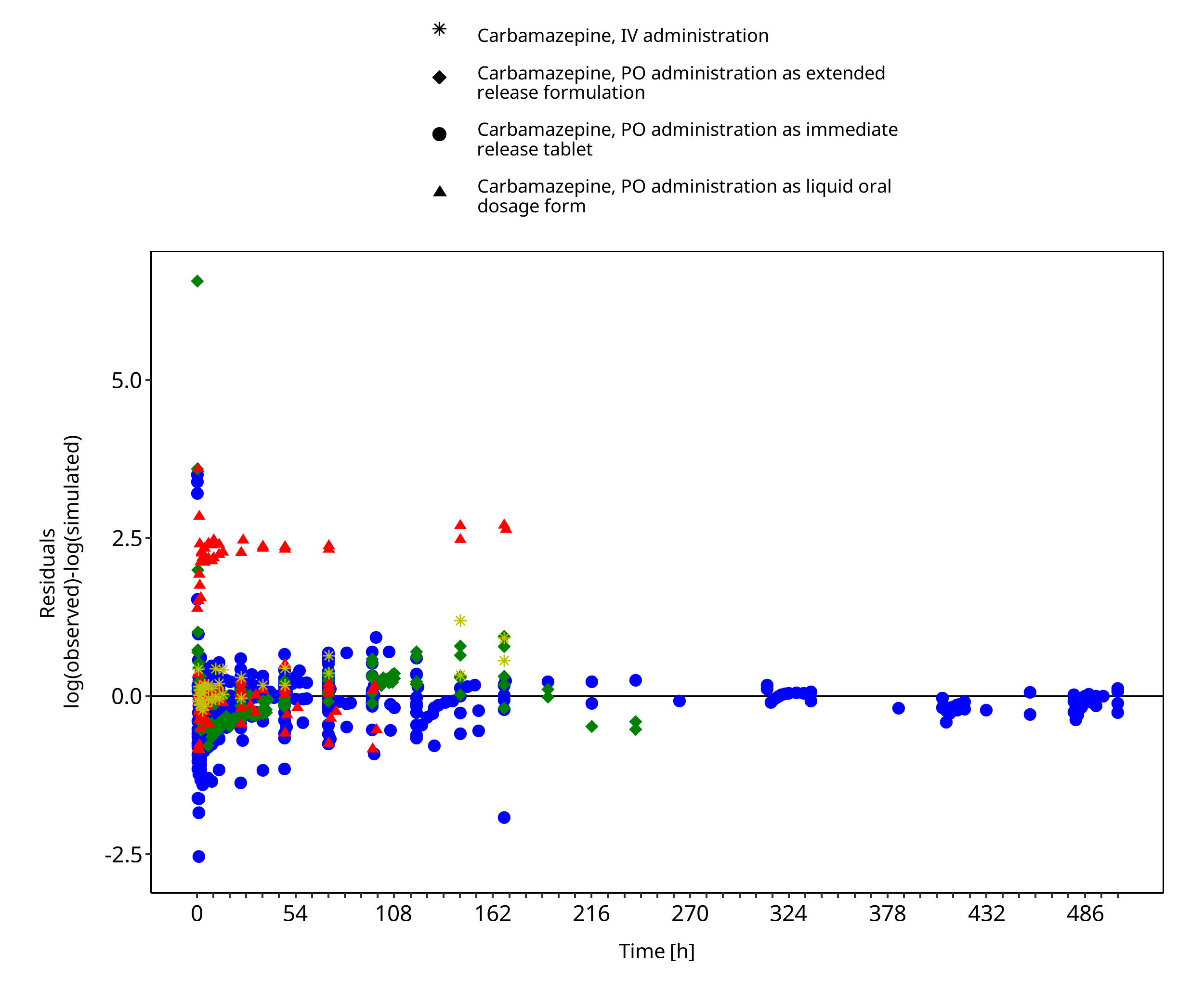
Figure 3-2: Goodness of fit plot for concentration in plasma
3.3 Concentration-Time Profiles¶
Simulated versus observed concentration-time profiles of all data listed in Section 2.2.2 are presented below.
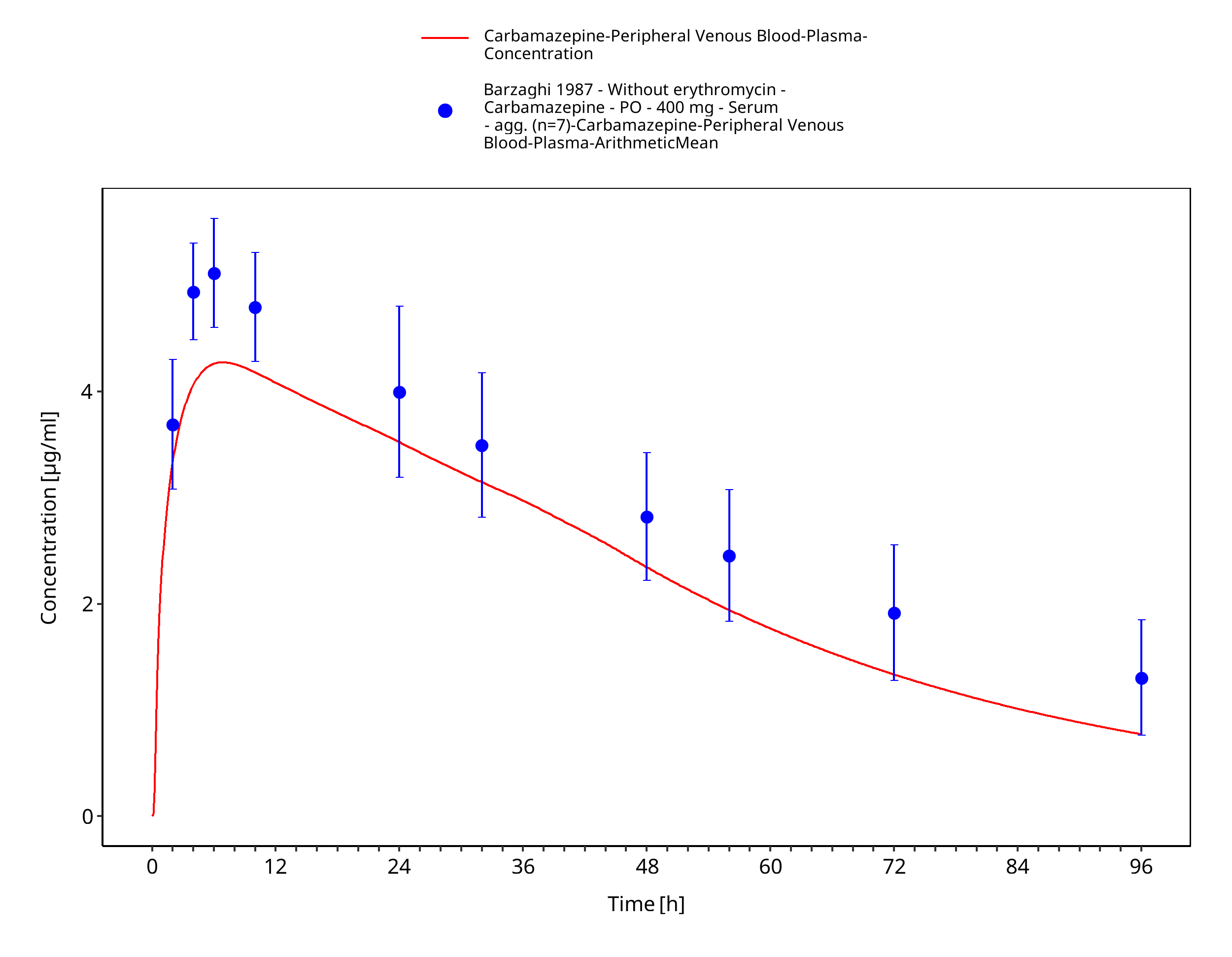
Figure 3-3: Barzaghi1987_400mg_sd_tabIR
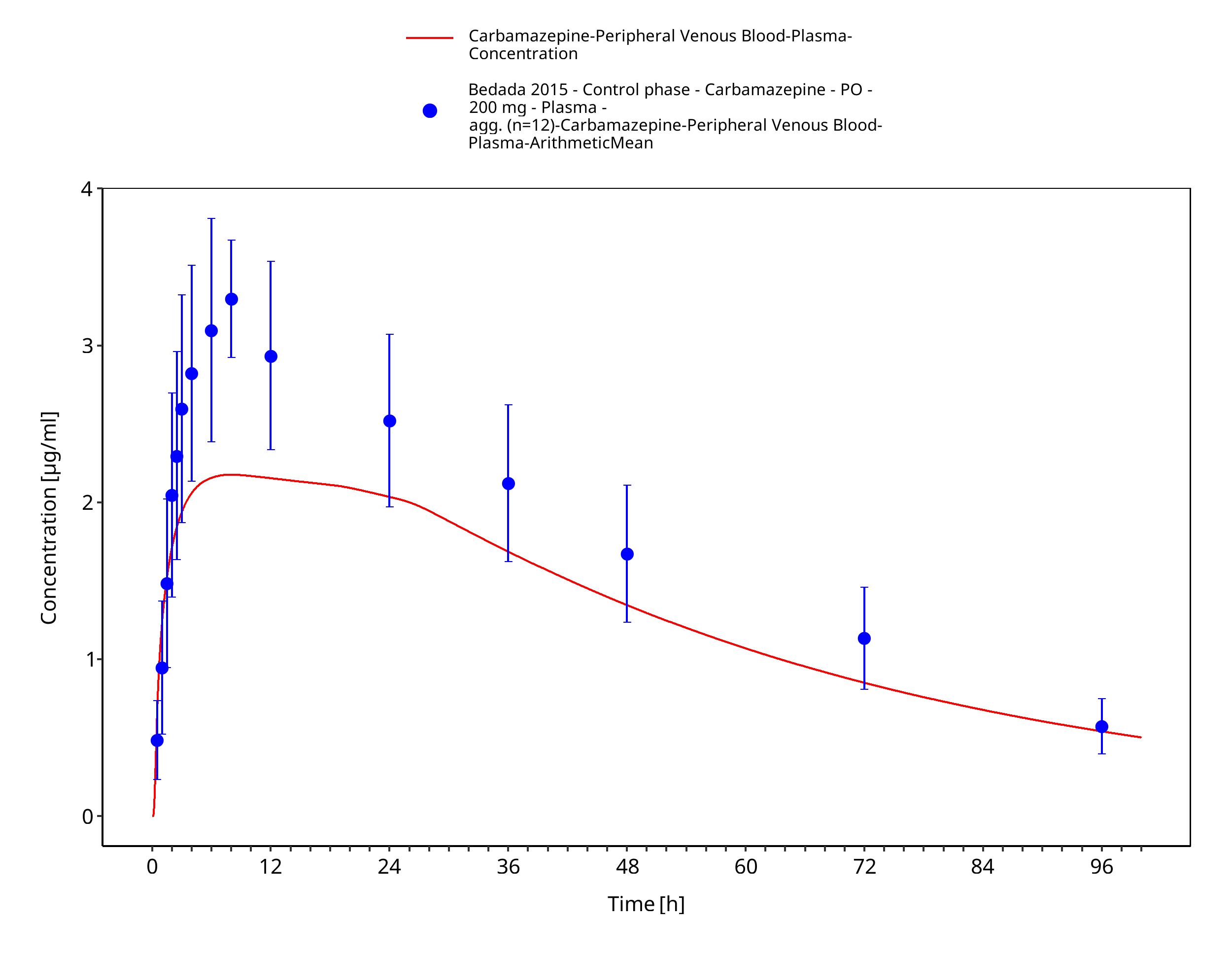
Figure 3-4: Bedada2015_200mg_sd_tabIR
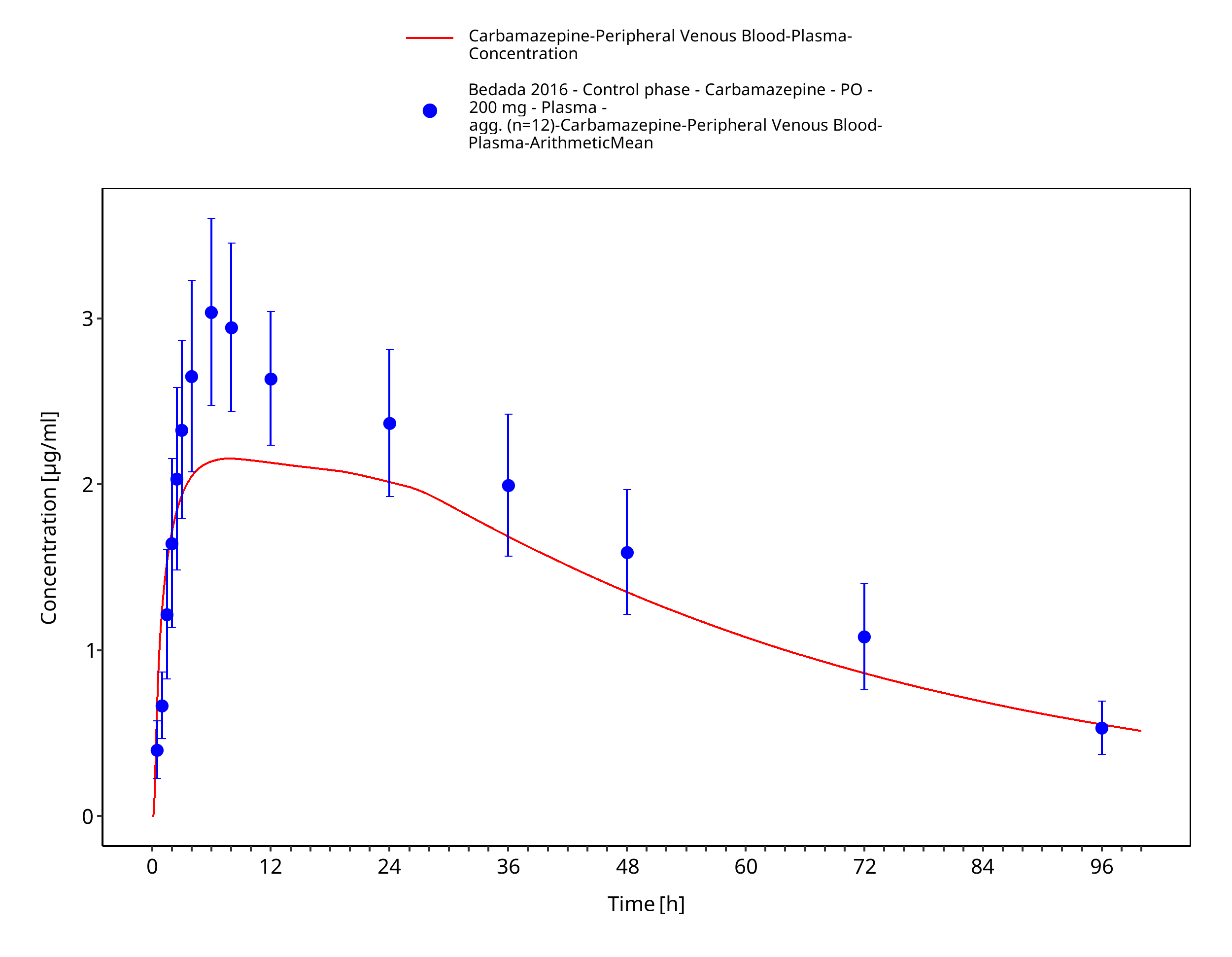
Figure 3-5: Bedada2016_200mg_sd_tabIR
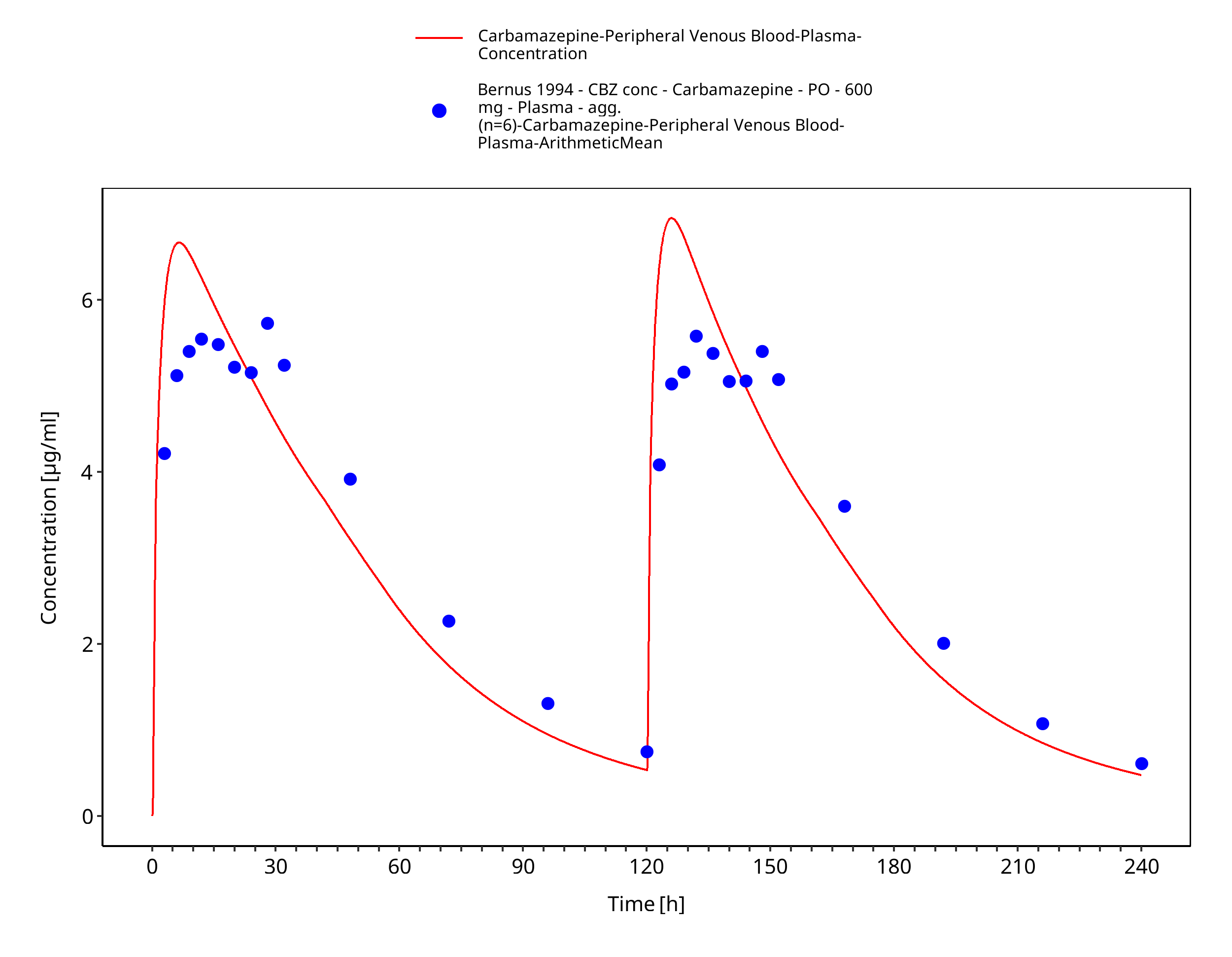
Figure 3-6: Bernus1994_600mg_D1+D5_tabIR - plasma
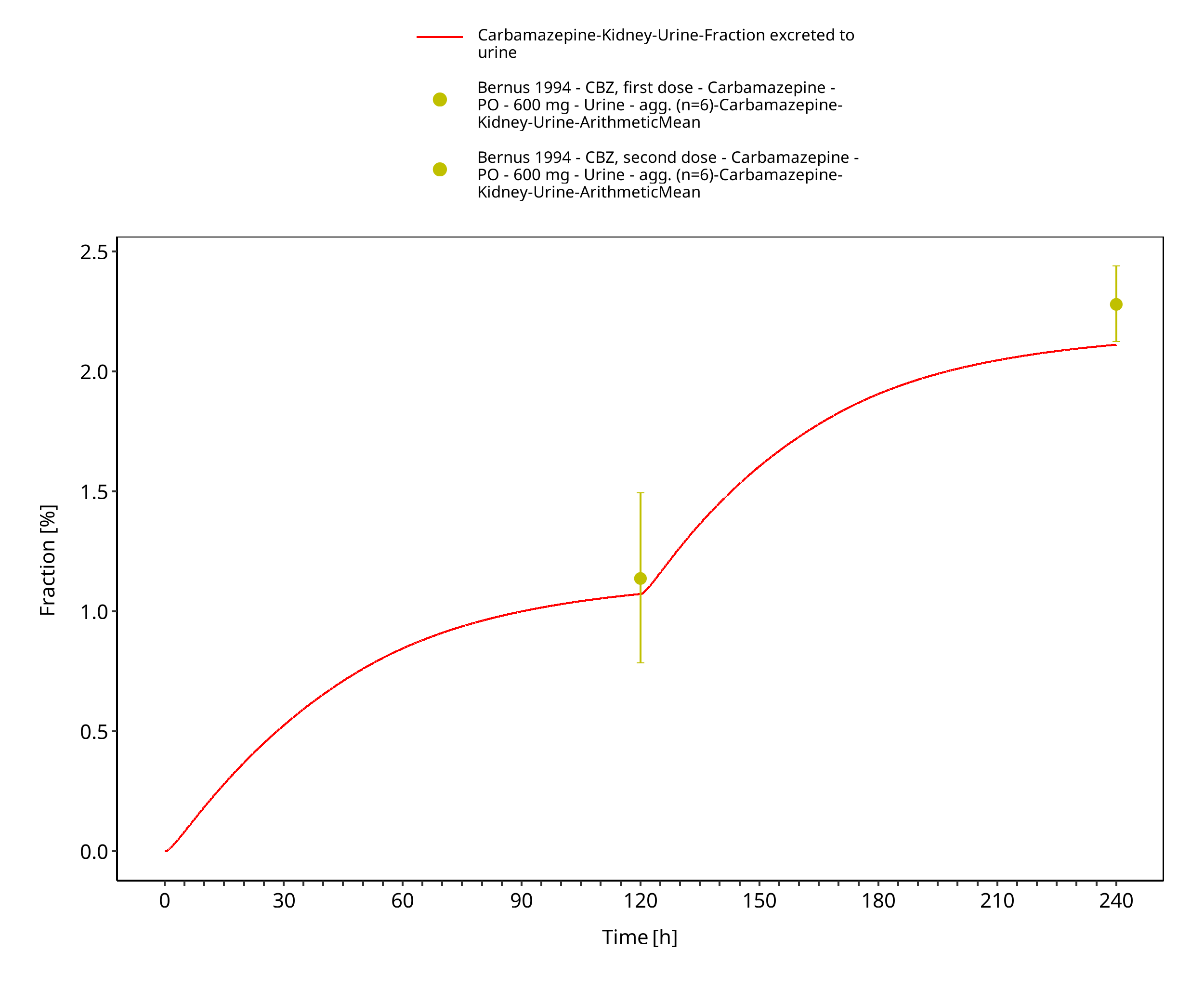
Figure 3-7: Bernus1994_600mg_D1+D5_tabIR - urine
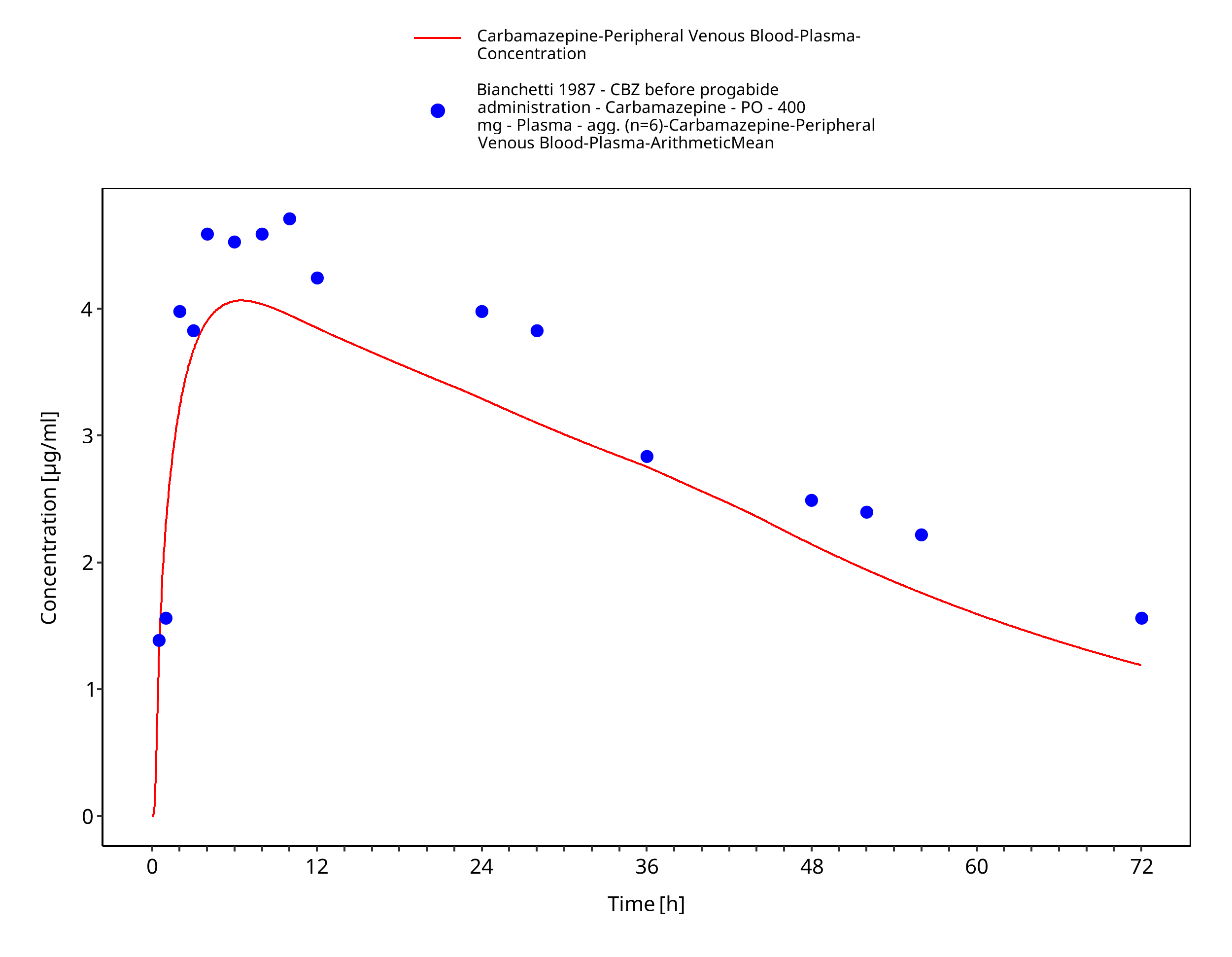
Figure 3-8: Bianchetti1987_400mg_sd_tabIR_fed
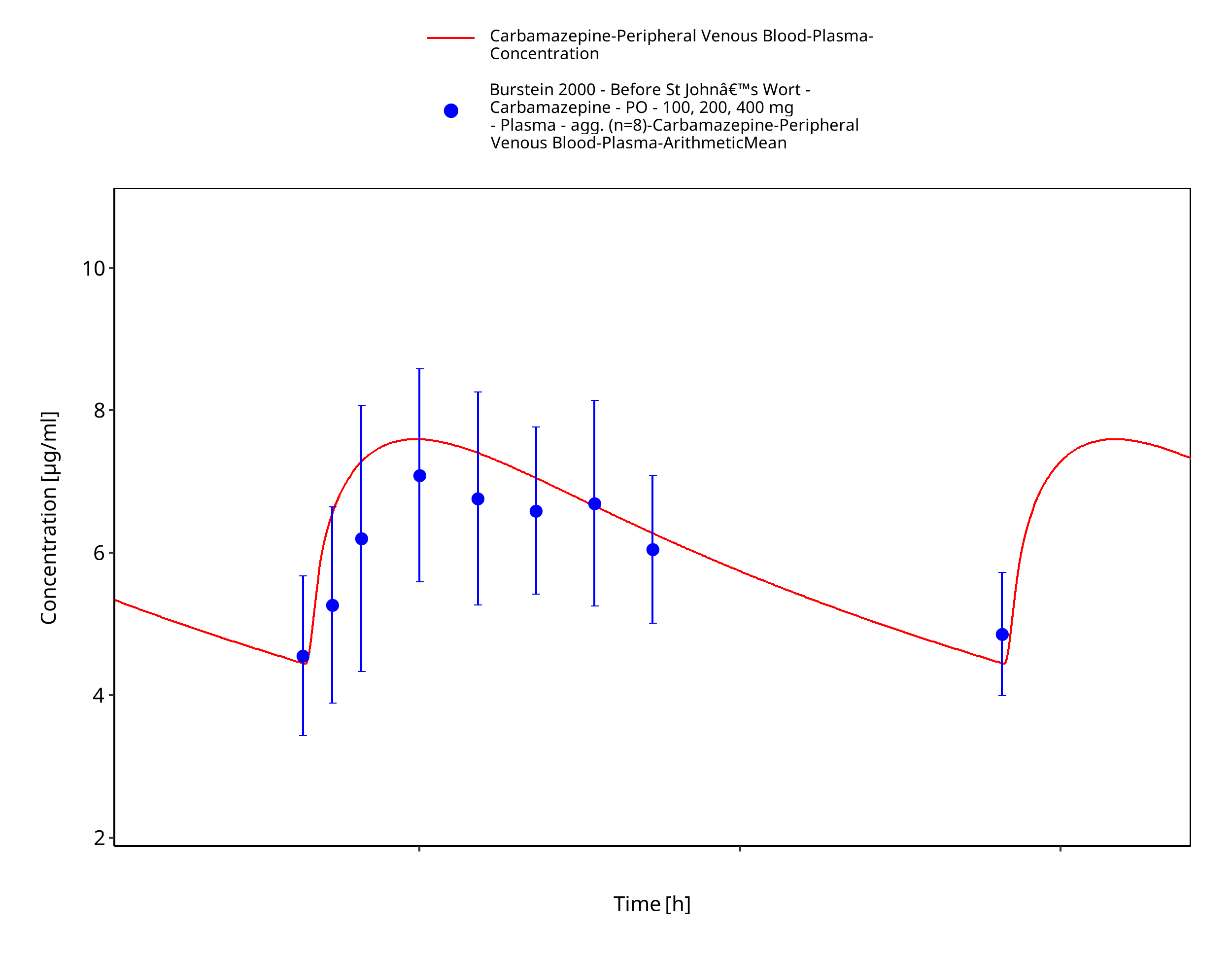
Figure 3-9: Burstein2000_100-200-400mg_md_tabIR
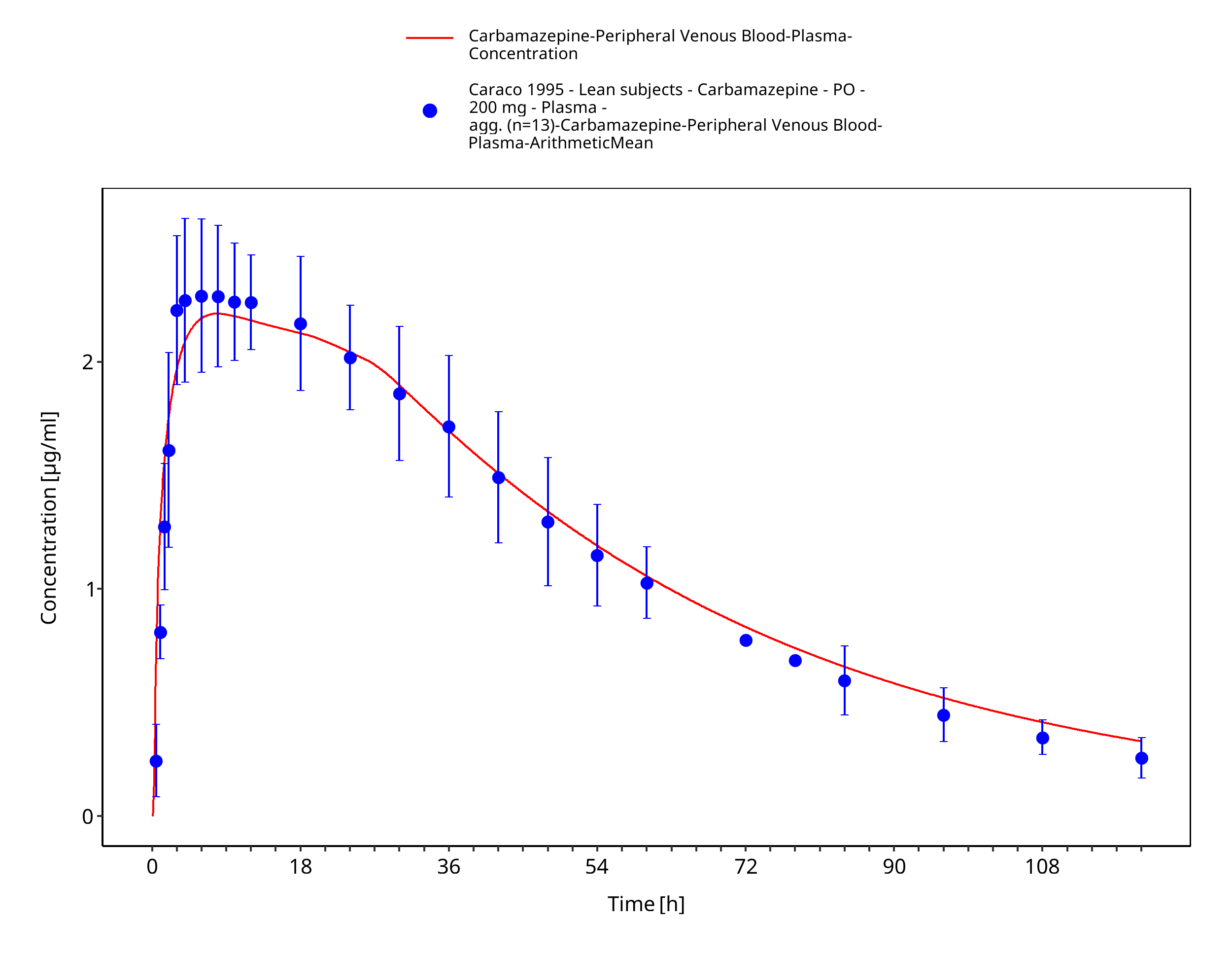
Figure 3-10: Time Profile Analysis
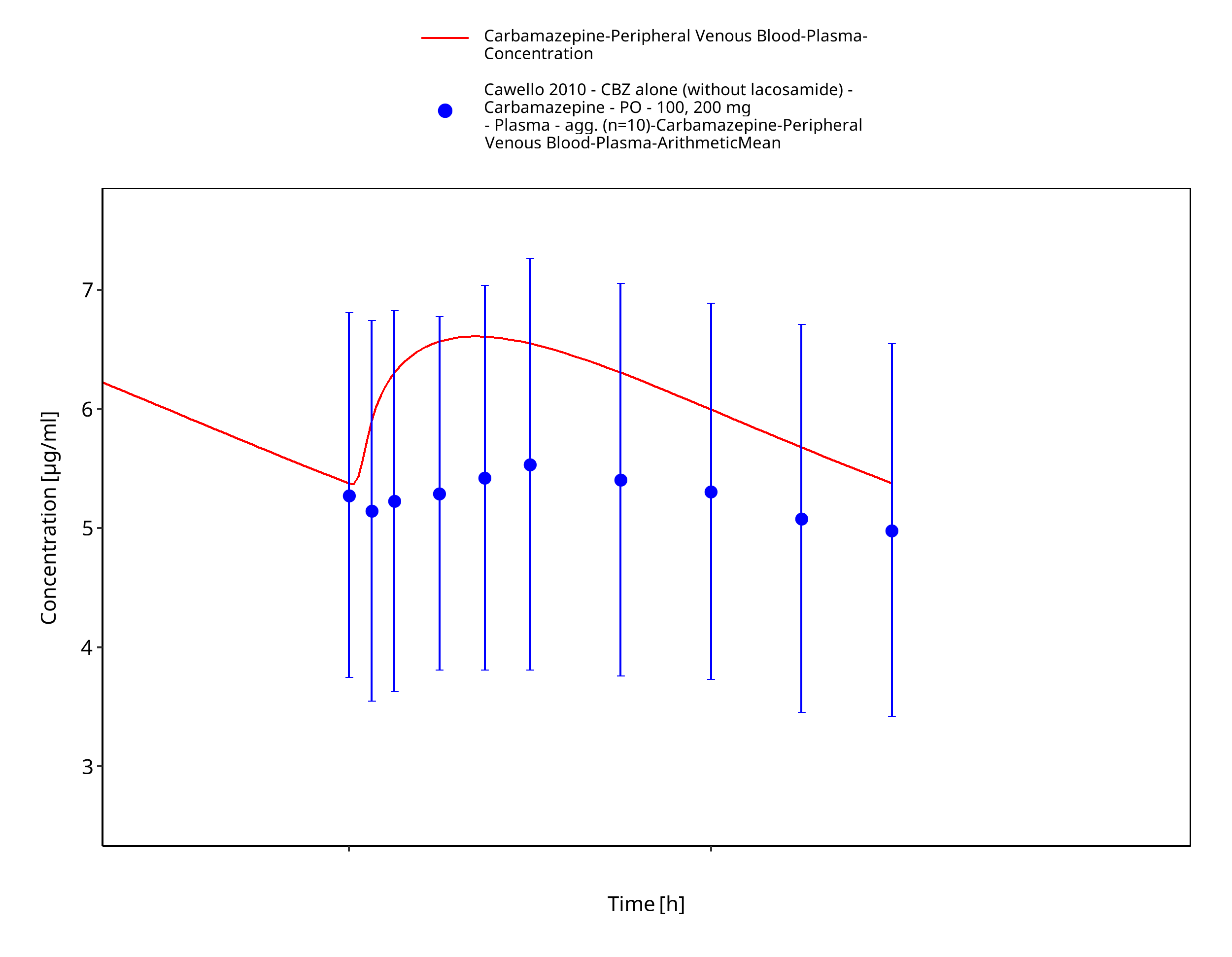
Figure 3-11: Time Profile Analysis
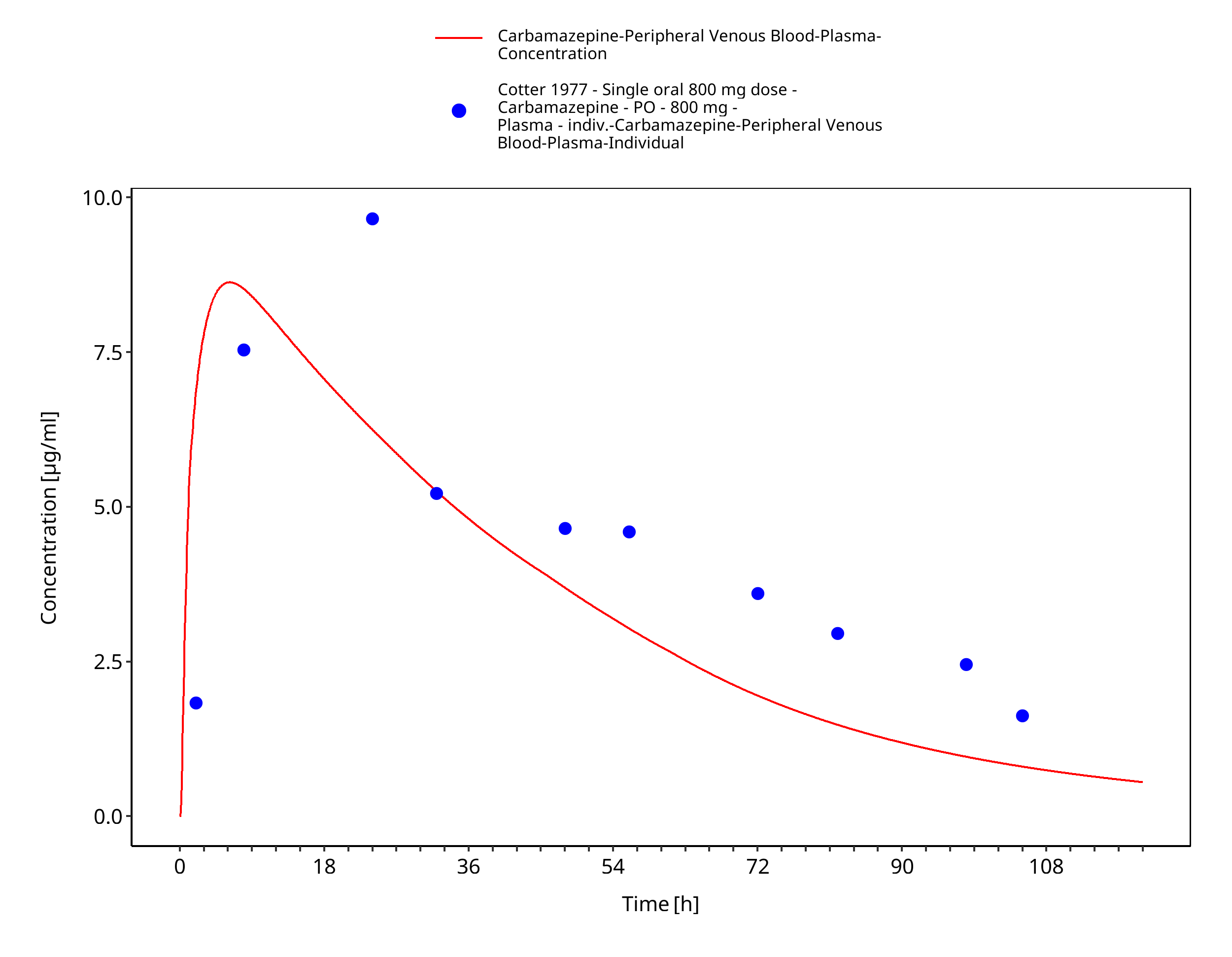
Figure 3-12: Cotter1977_individual_800mg_sd_tabIR
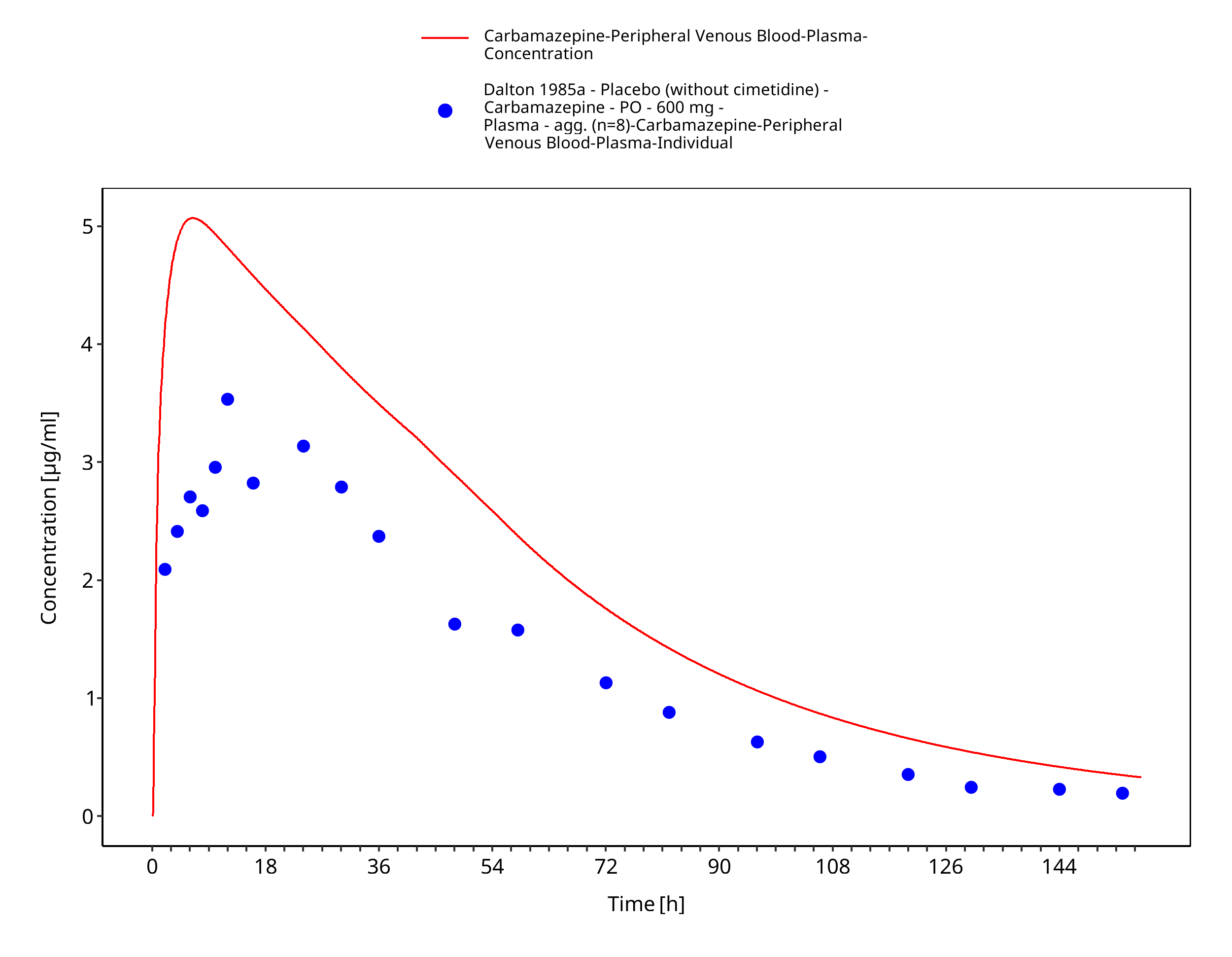
Figure 3-13: Dalton1985_control_600mg_sd_tabIR
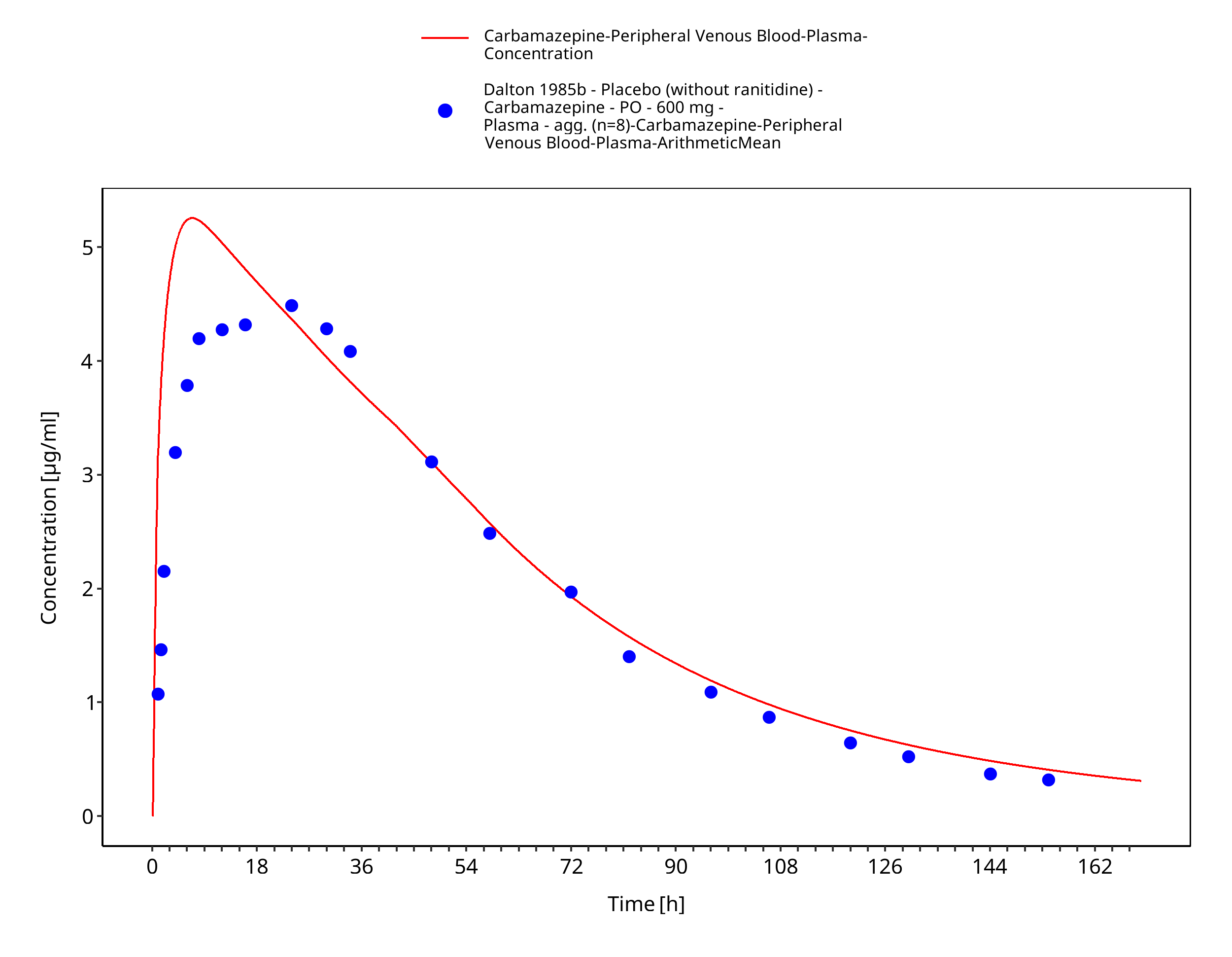
Figure 3-14: Dalton1985a_600mg_sd_tabIR
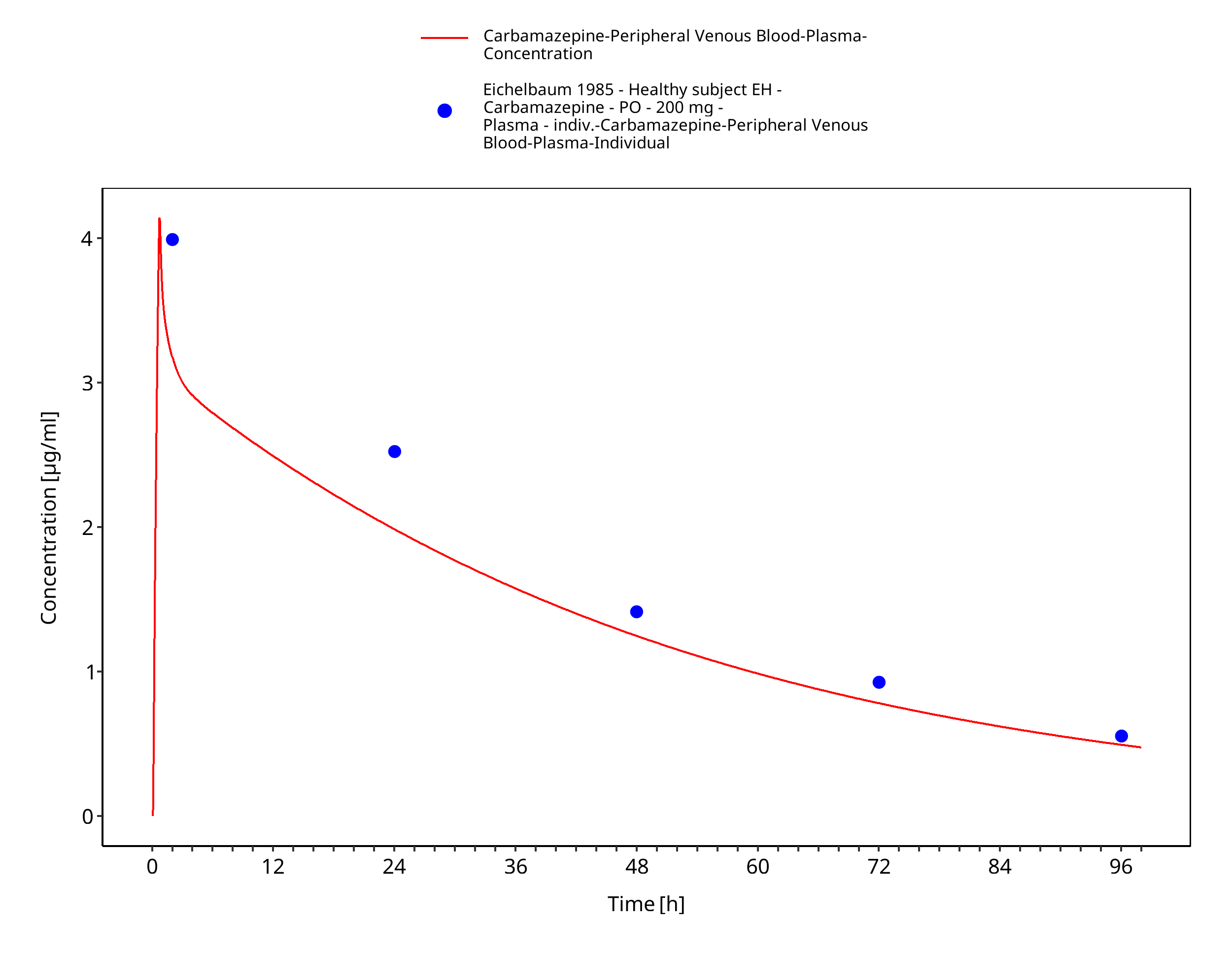
Figure 3-15: Time Profile Analysis
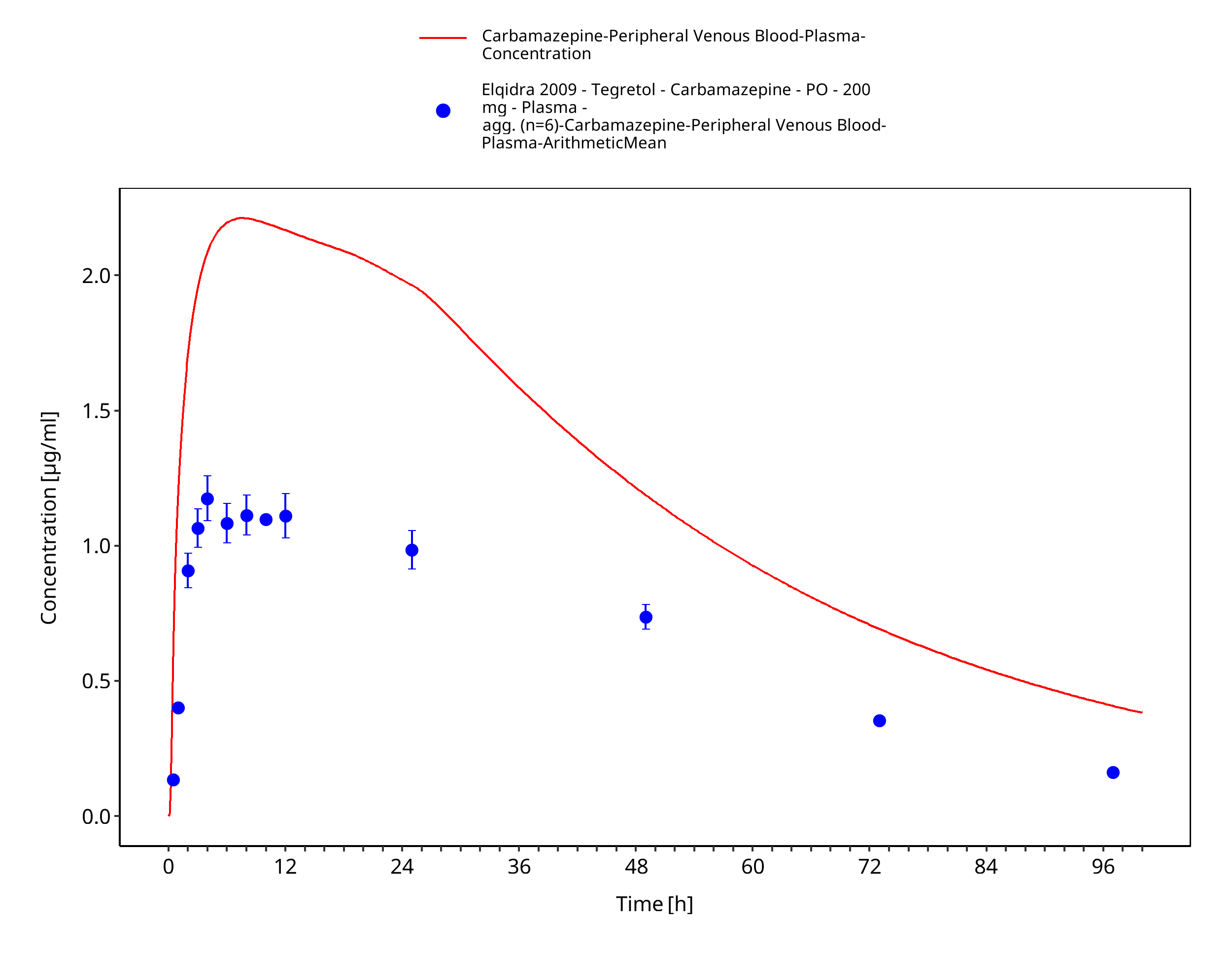
Figure 3-16: Time Profile Analysis
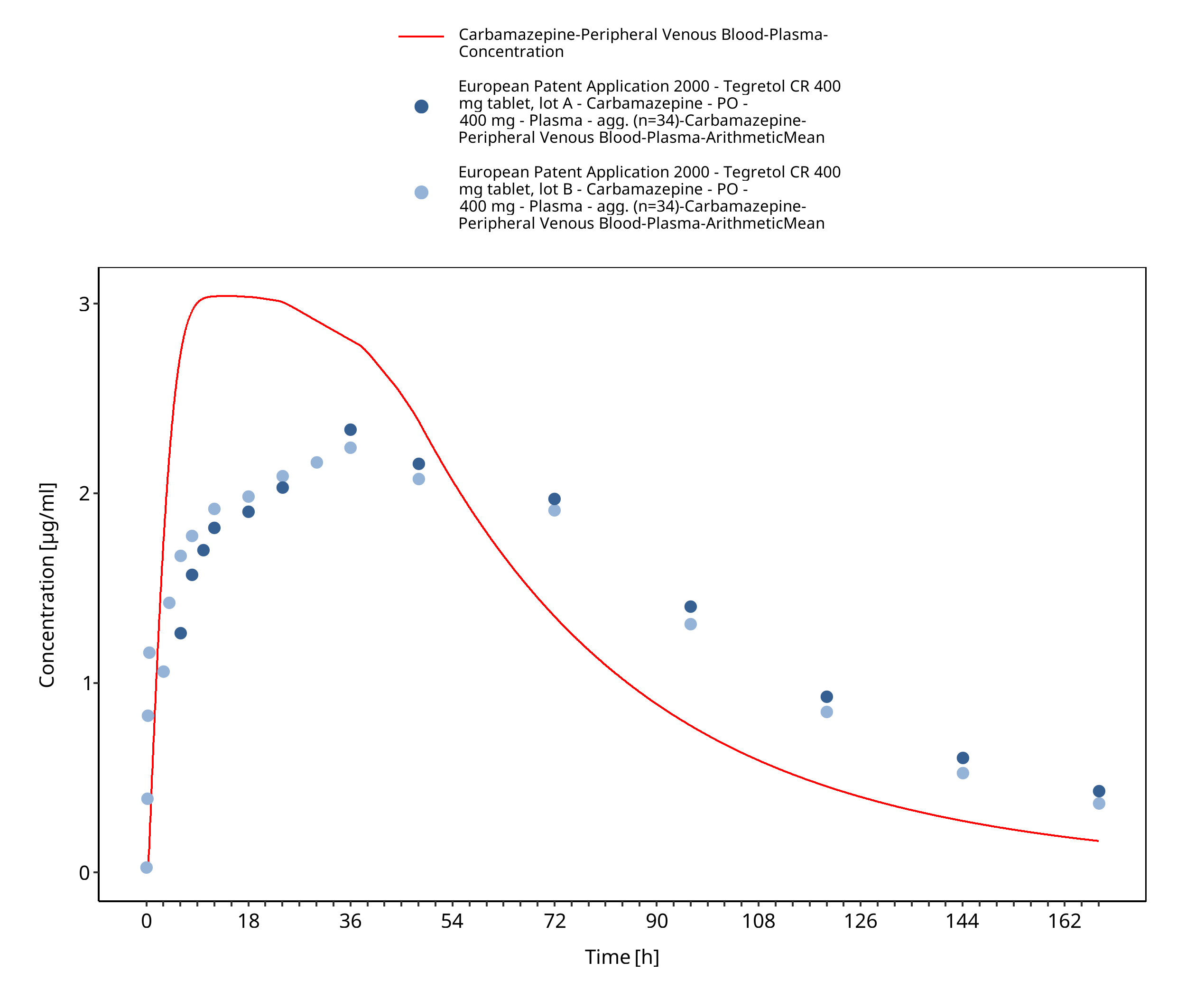
Figure 3-17: EUPatent2005_400mg_sd_TegretolXR
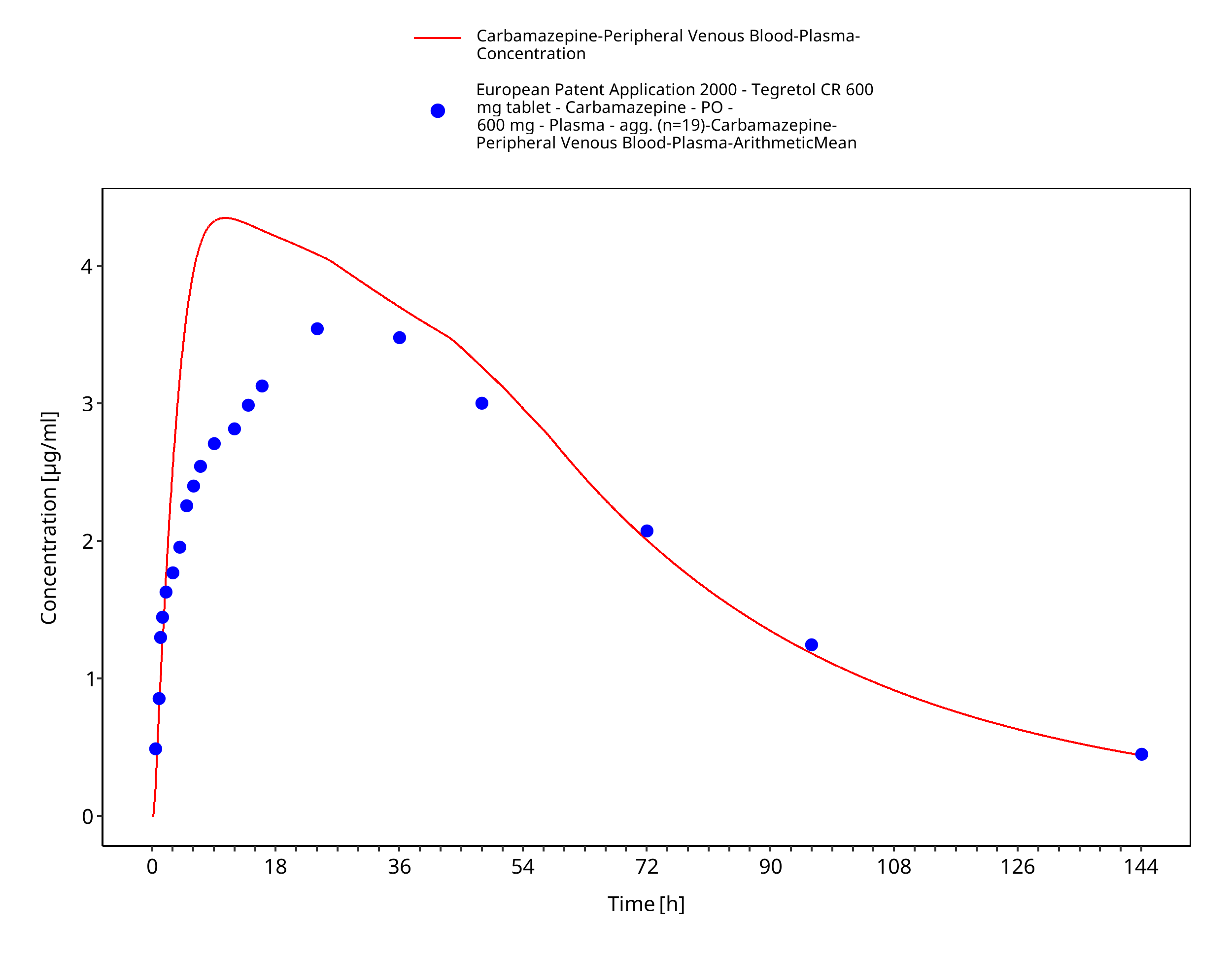
Figure 3-18: EUPatent2005_600mg_sd_TegretolXR
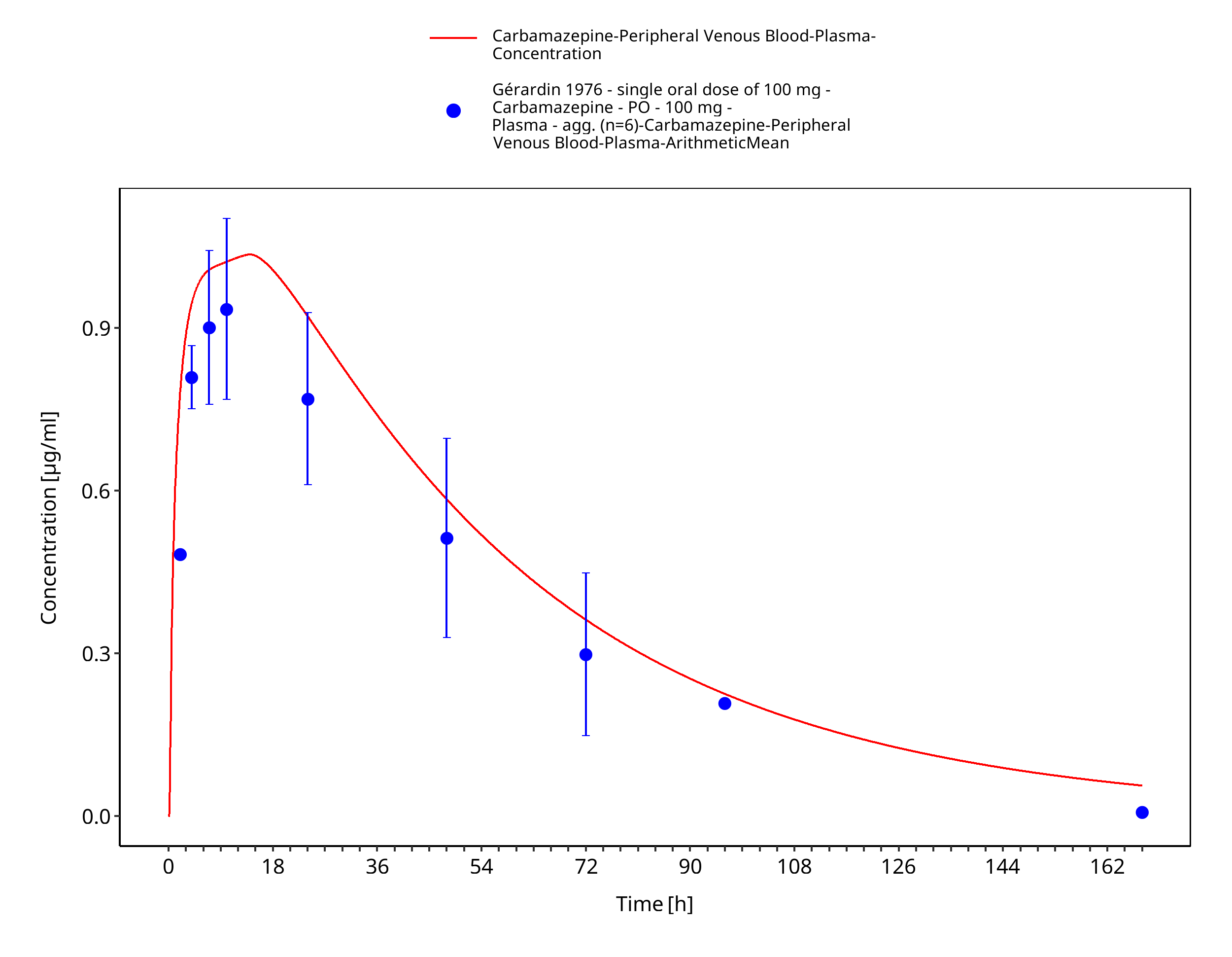
Figure 3-19: Time Profile Analysis
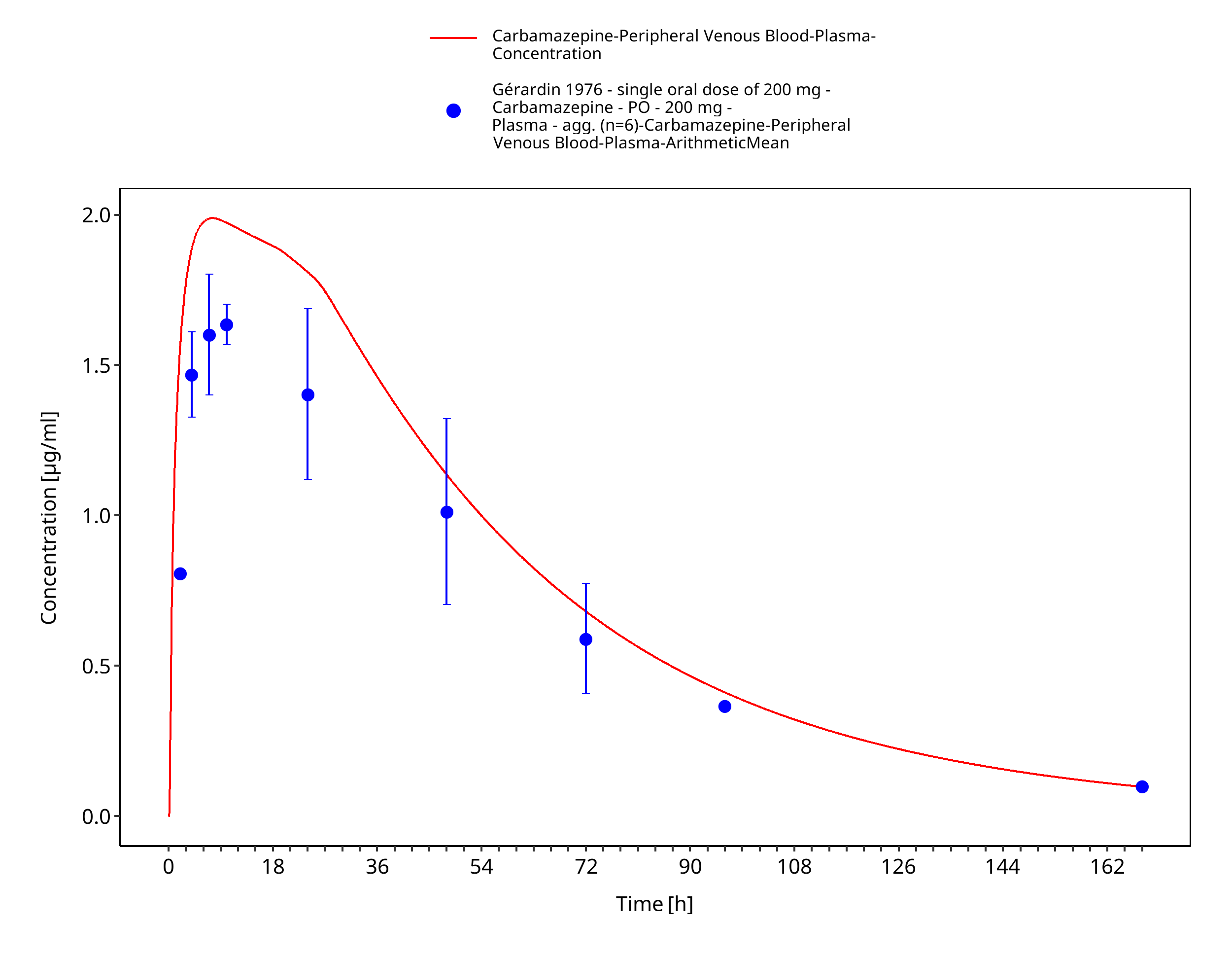
Figure 3-20: Geradin1976_200mg_sd_tabIR
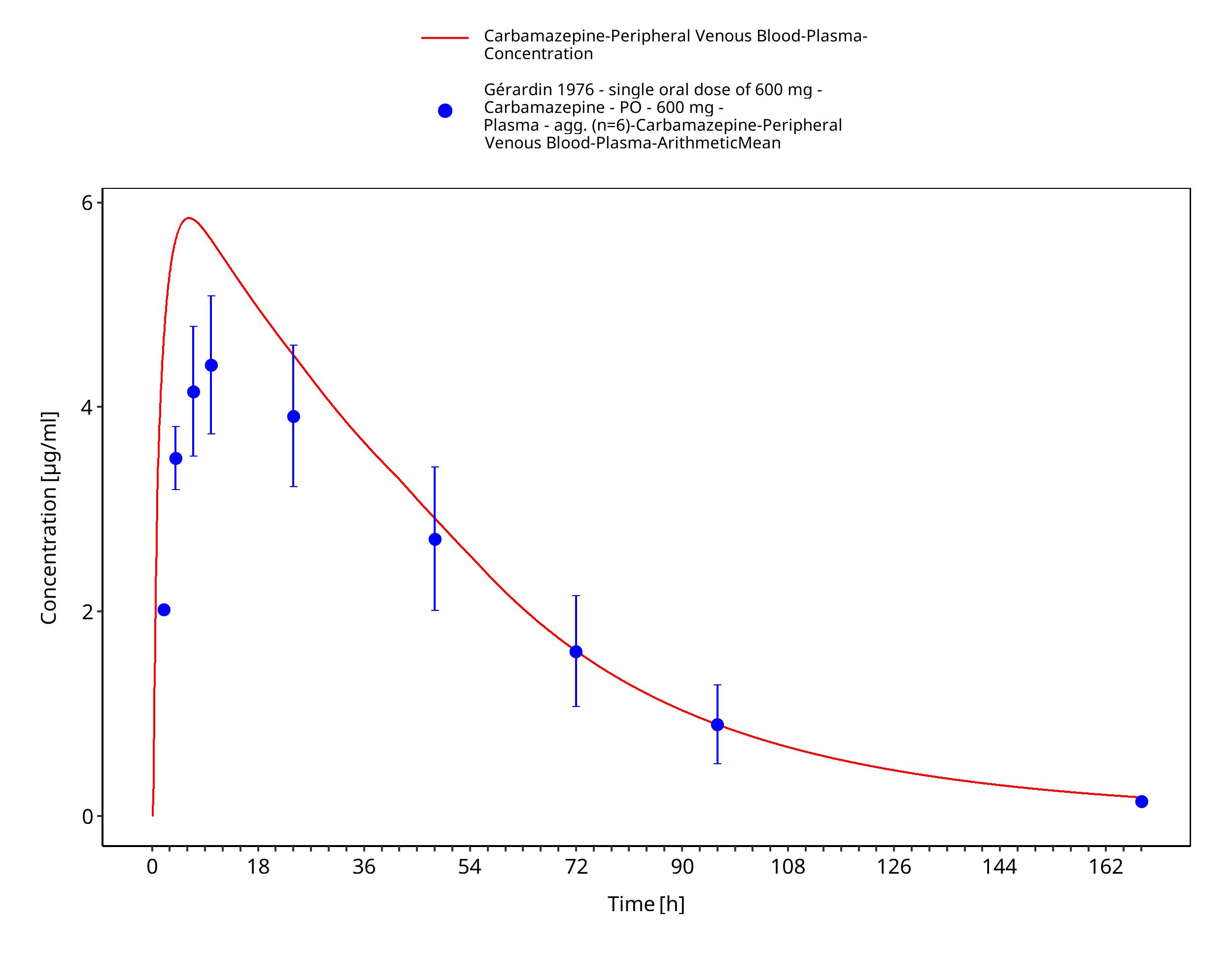
Figure 3-21: Time Profile Analysis
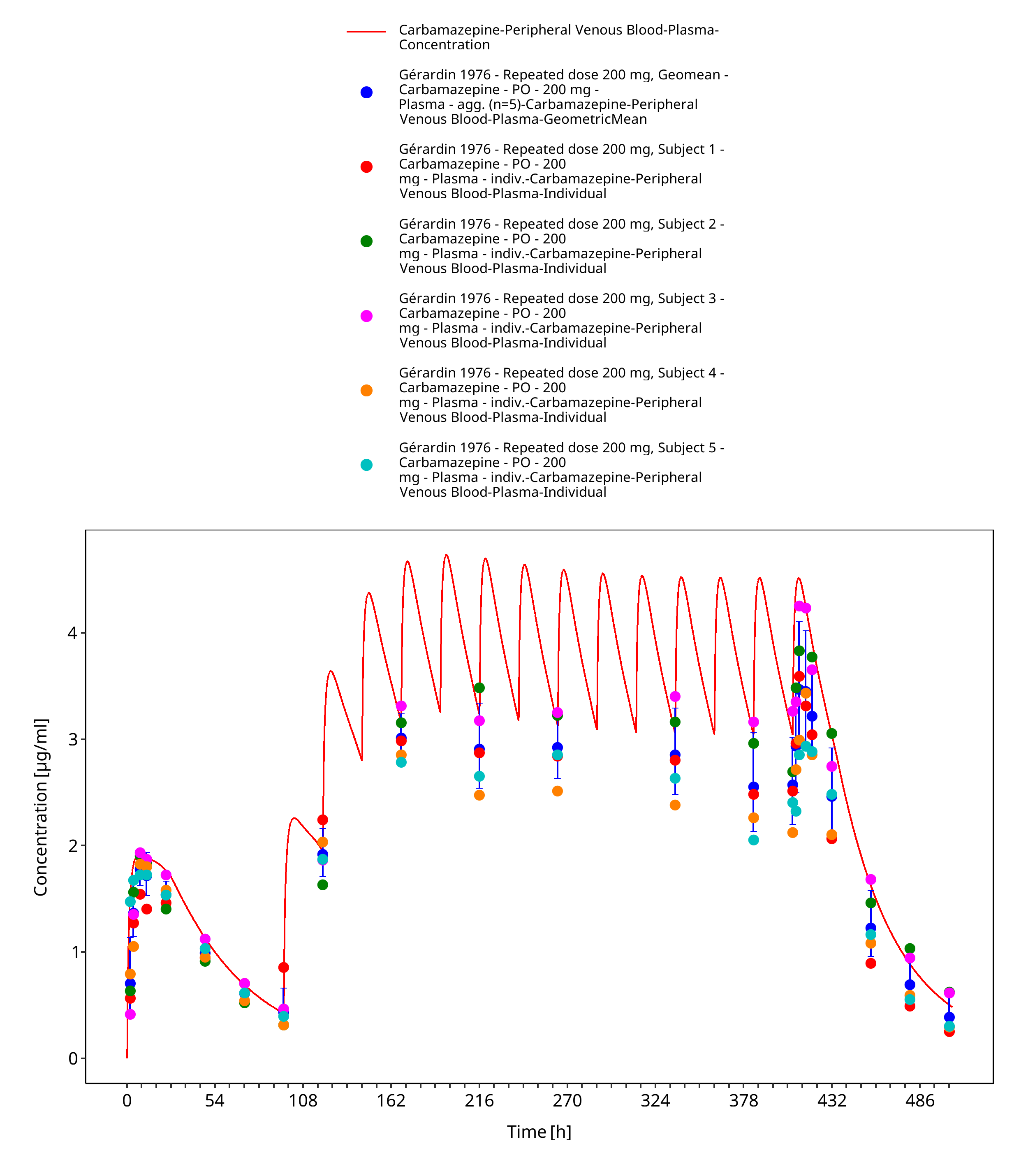
Figure 3-22: Geradin1976_mean_200mg_md_tabIR
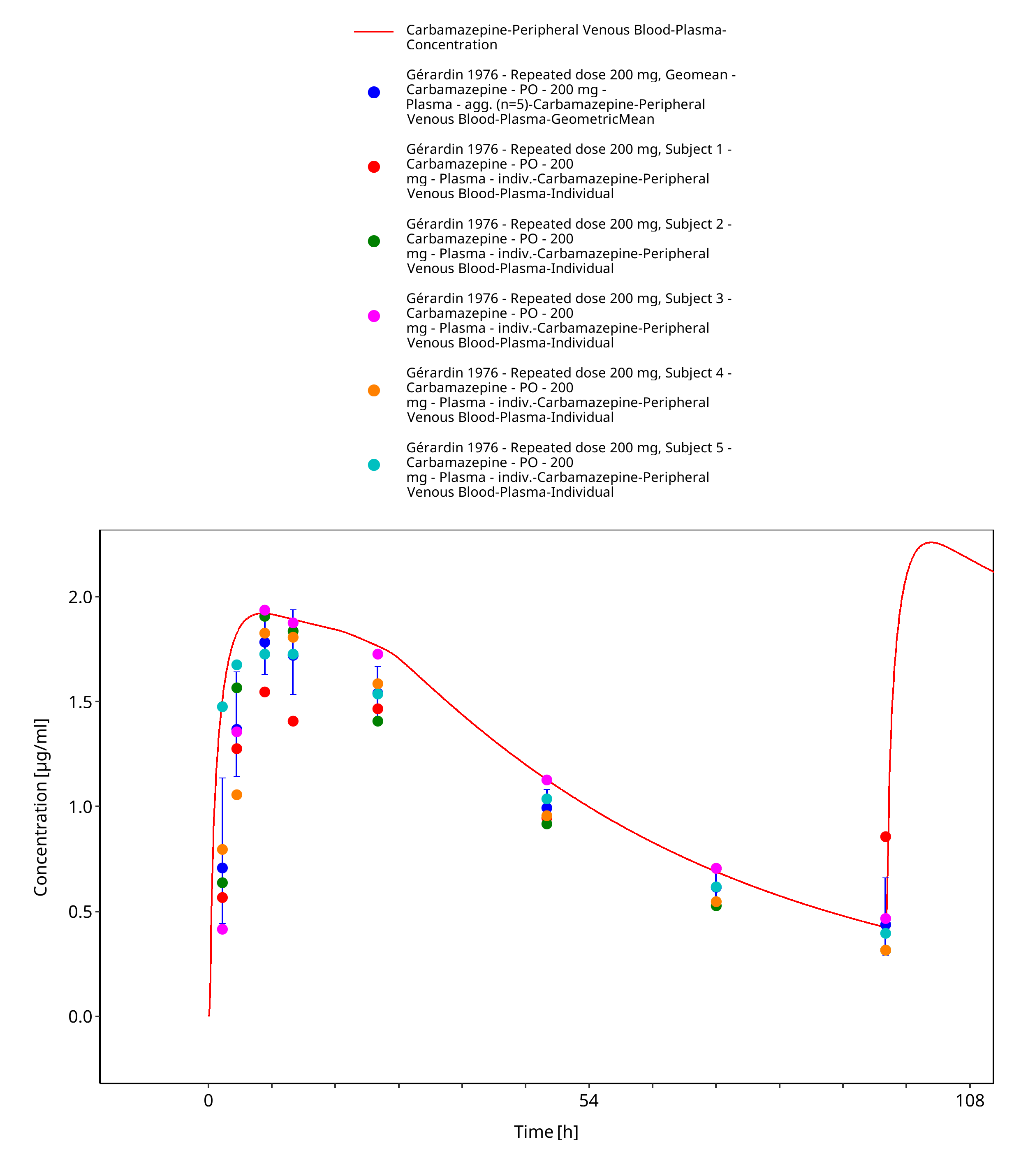
Figure 3-23: Geradin1976_mean_200mg_md_tabIR - first dose
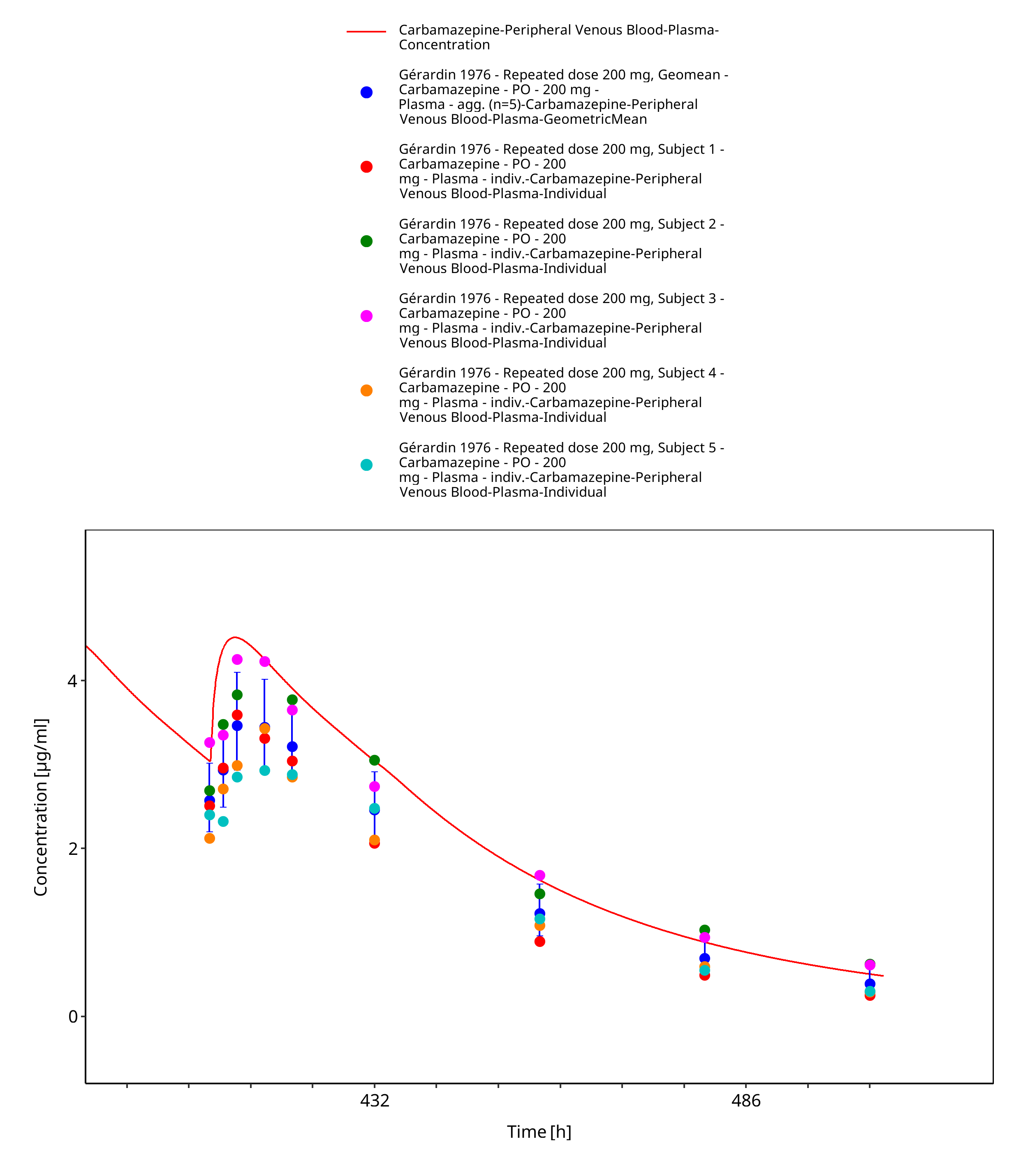
Figure 3-24: Geradin1976_mean_200mg_md_tabIR - last dose
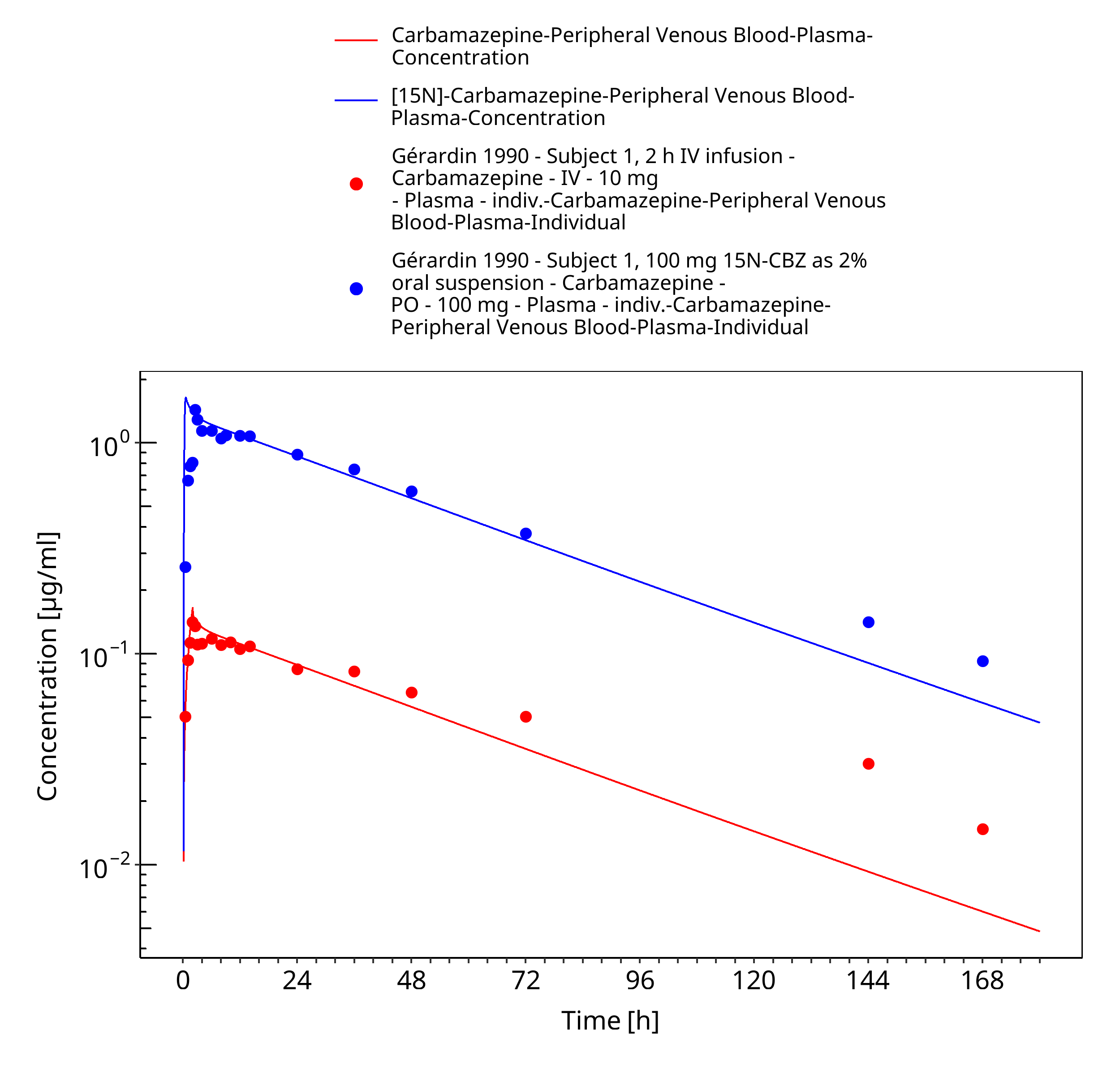
Figure 3-25: Geradin1990_Subject1_100mg-iv_100mg-po-sol
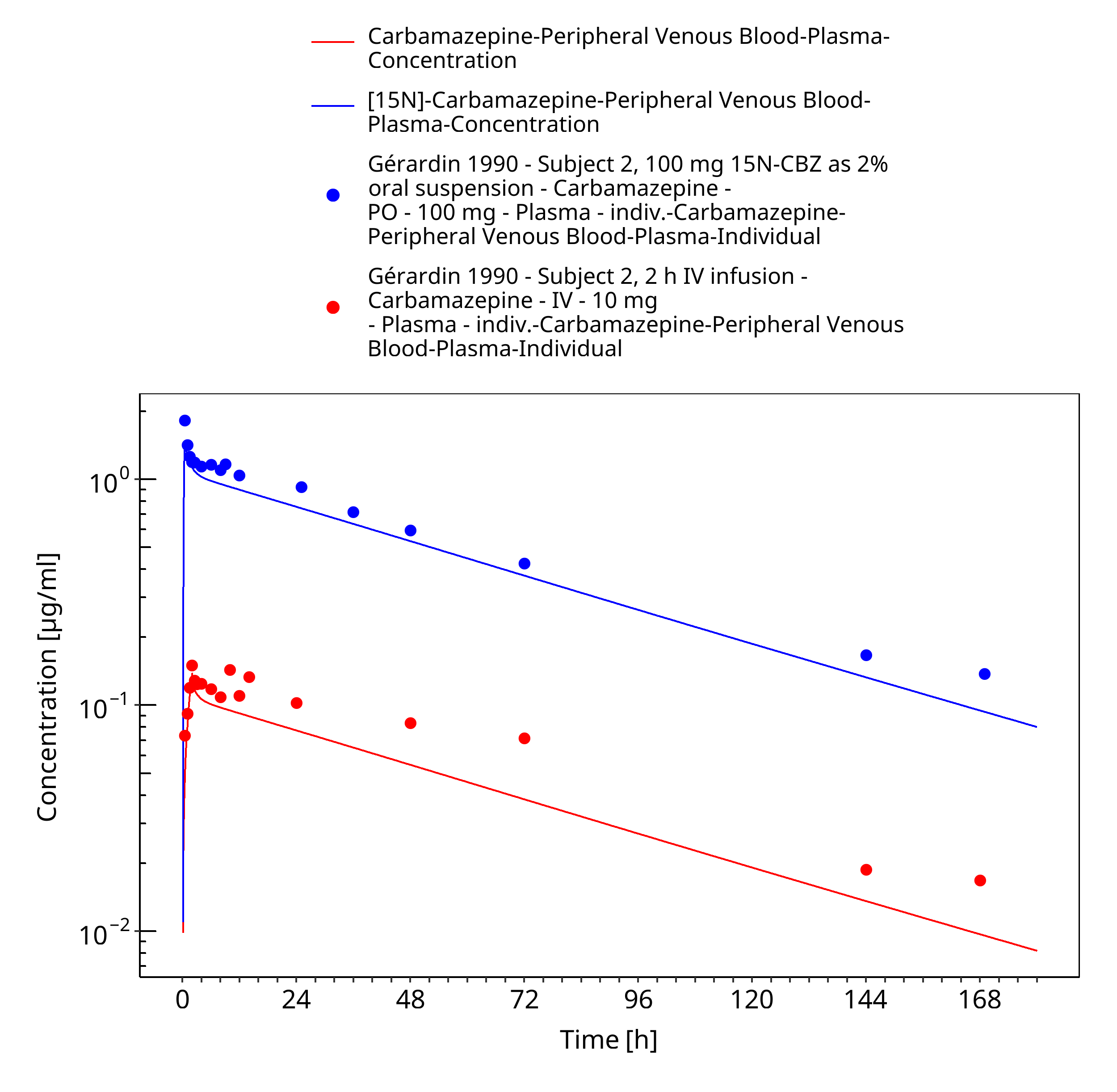
Figure 3-26: Geradin1990_Subject2_100mg-iv_100mg-po-sol
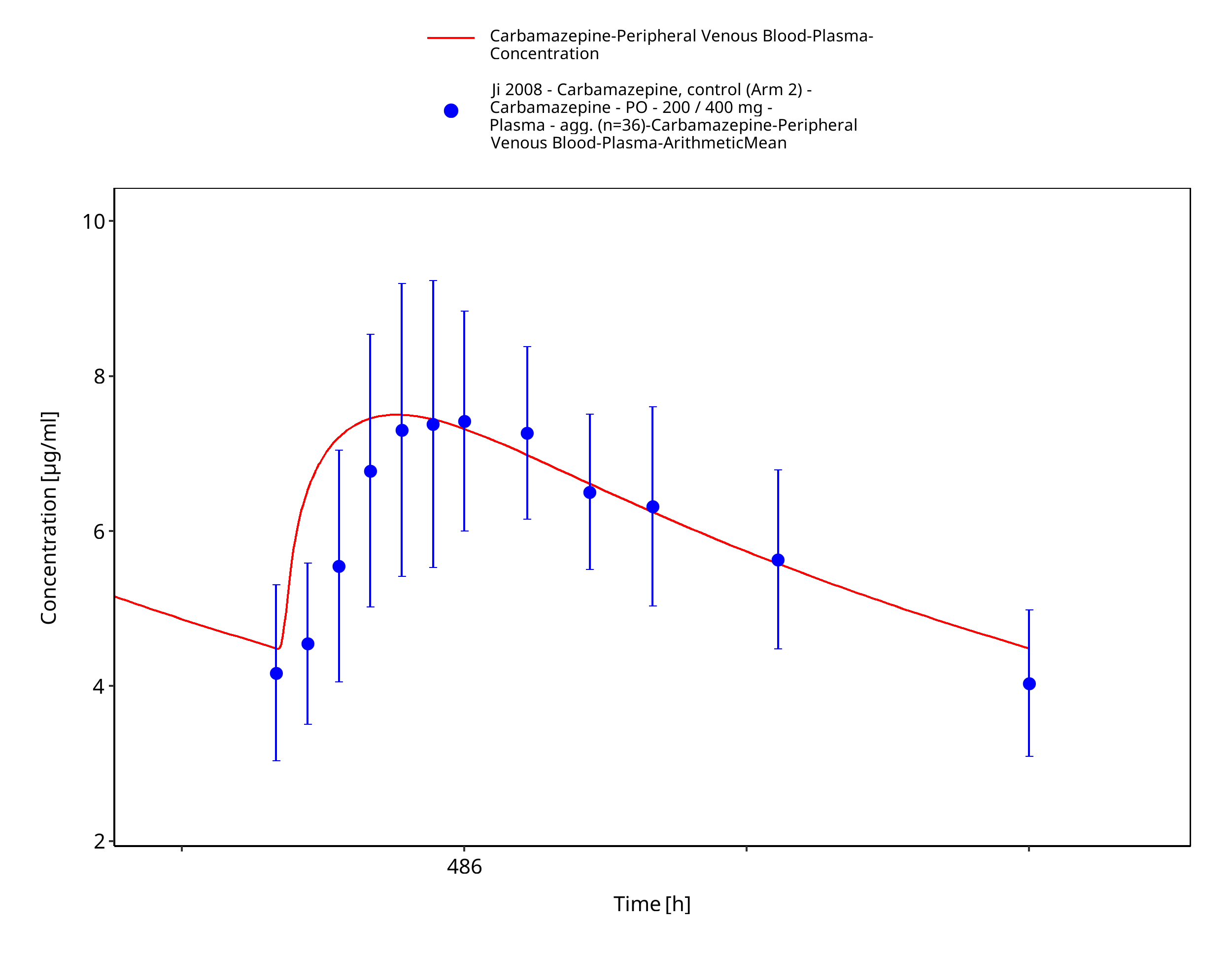
Figure 3-27: Ji2008_100-200-400mg_md_tabIR
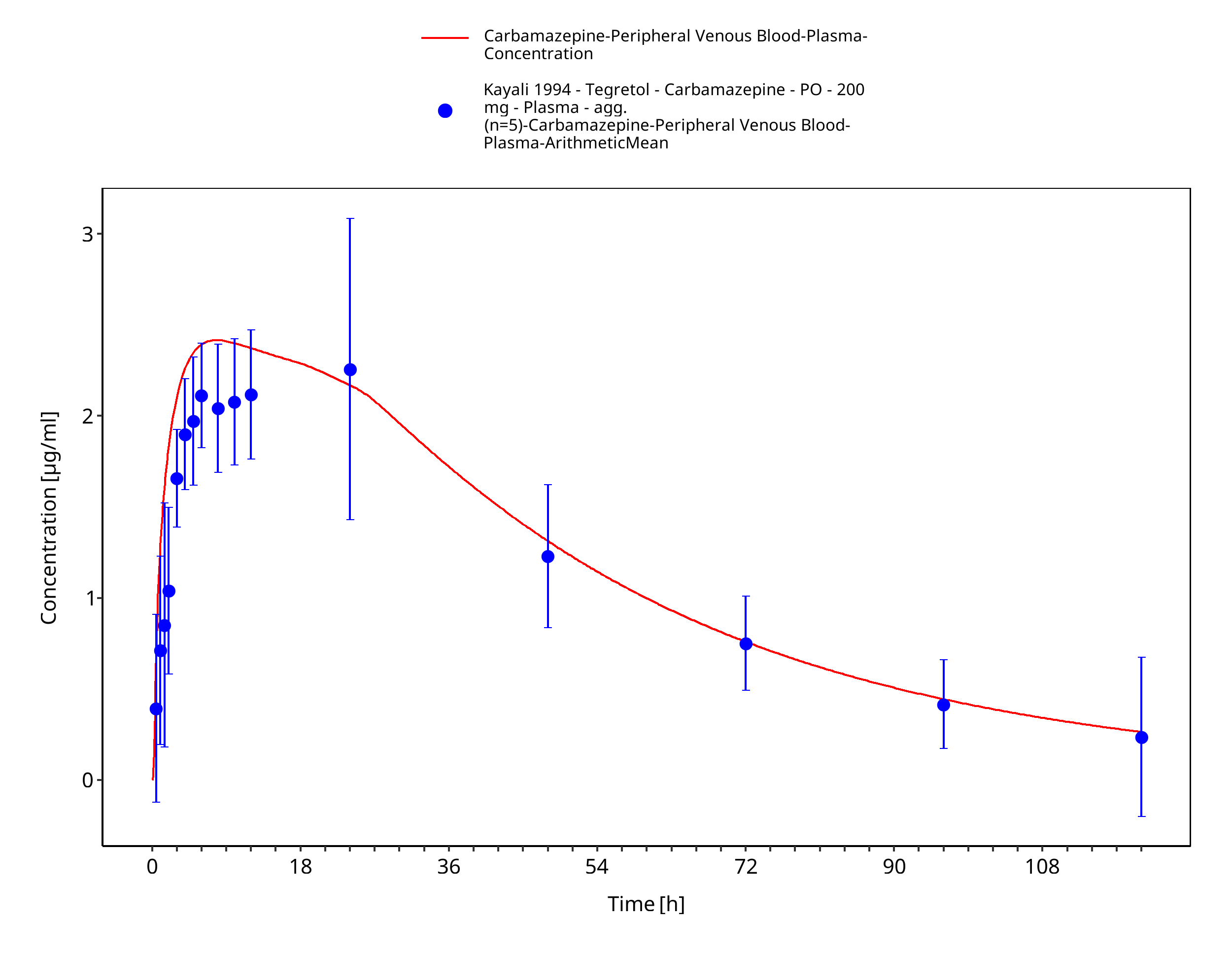
Figure 3-28: Time Profile Analysis
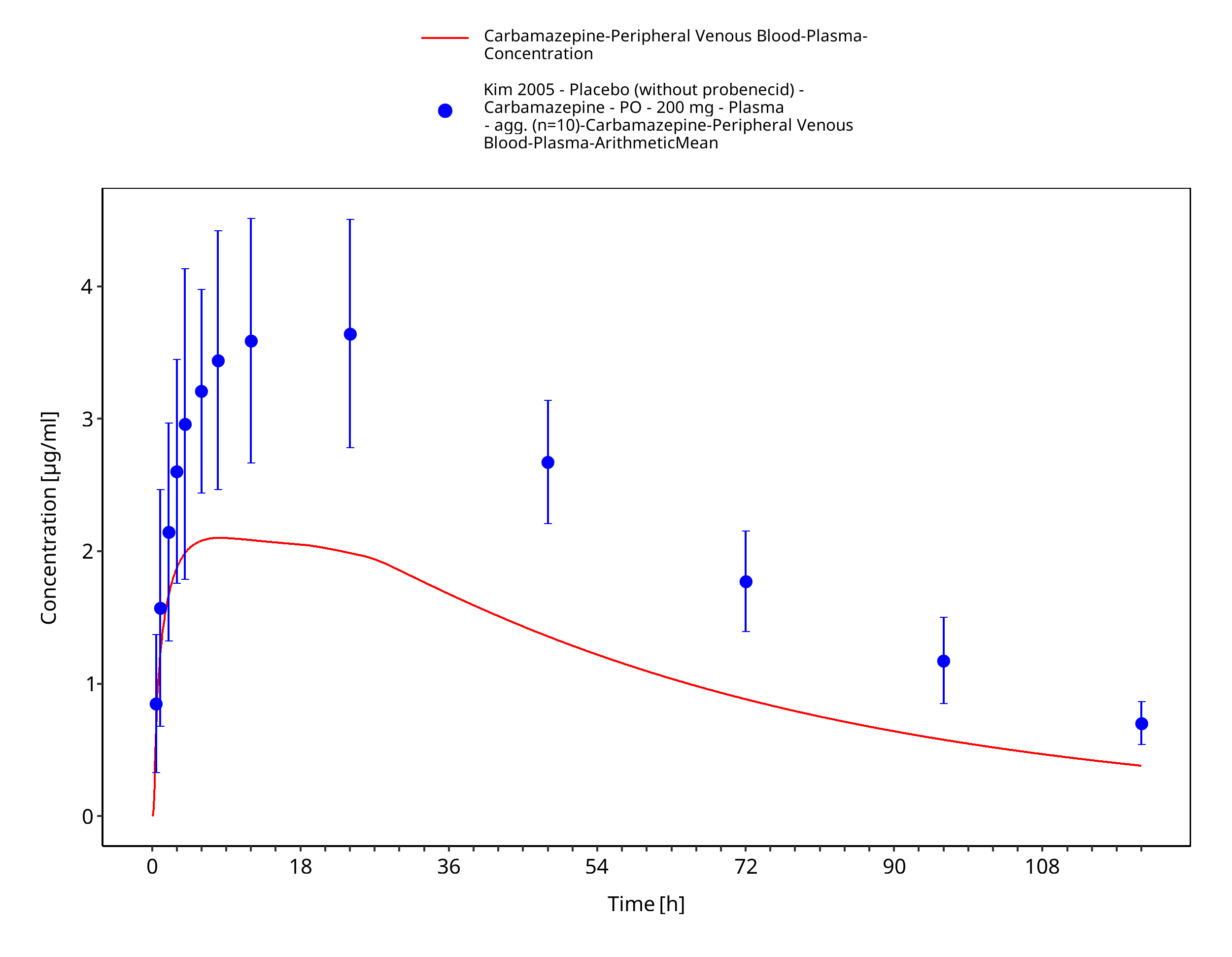
Figure 3-29: Kim2005_200mg_sd_tabIR
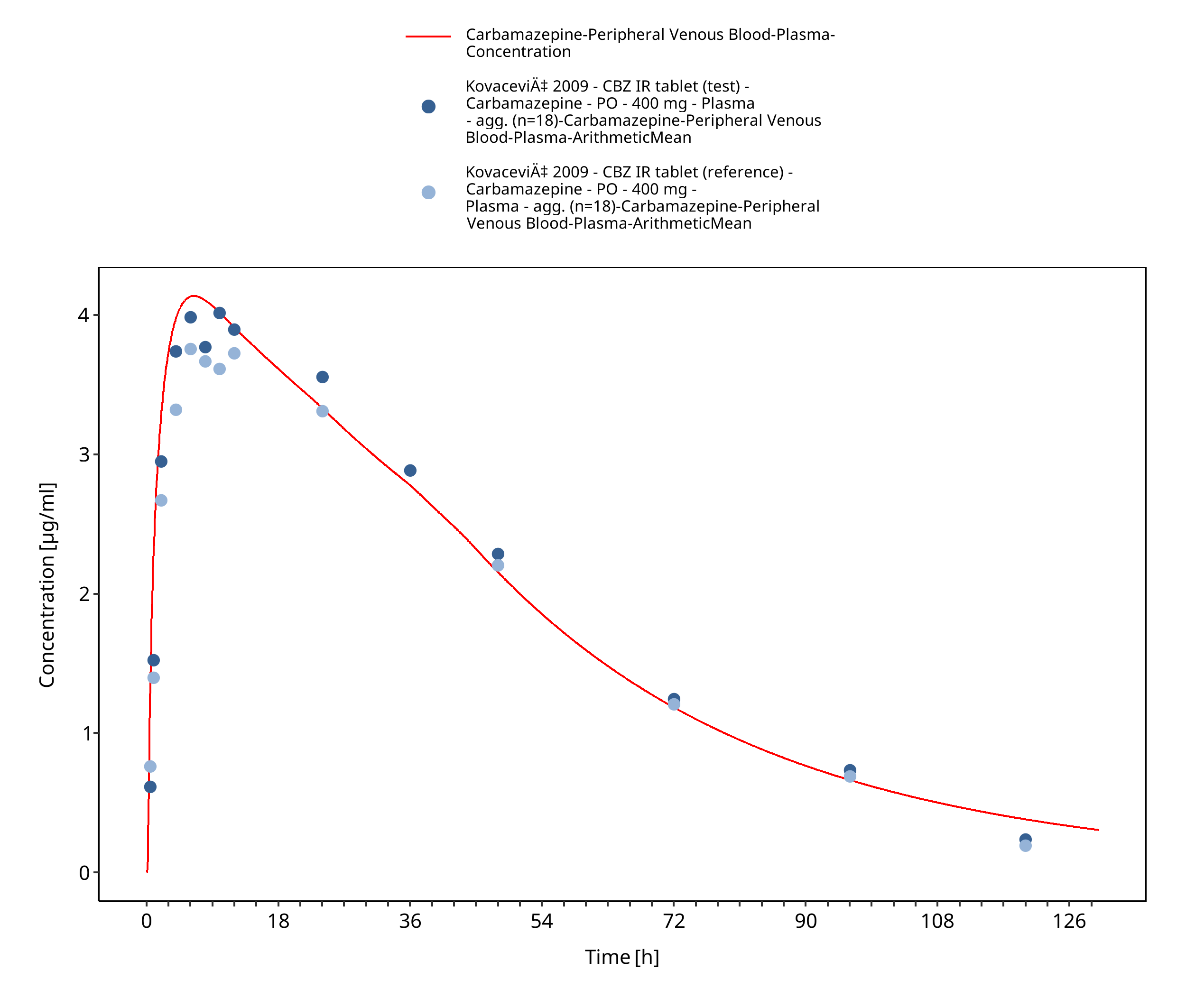
Figure 3-30: Kovacevic2009_400mg_sd_TegretolIR
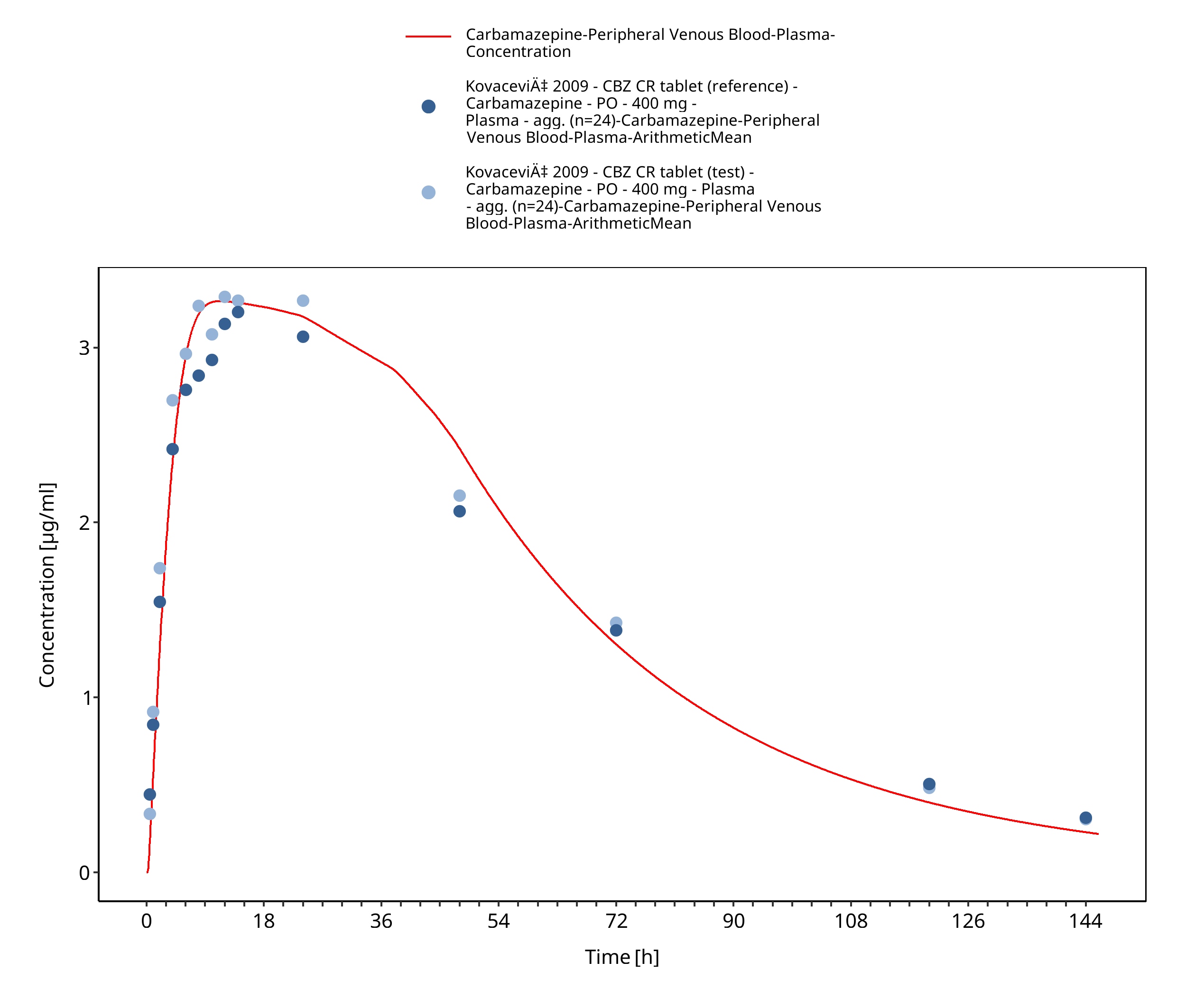
Figure 3-31: Kovacevic2009_400mg_sd_TegretolXR
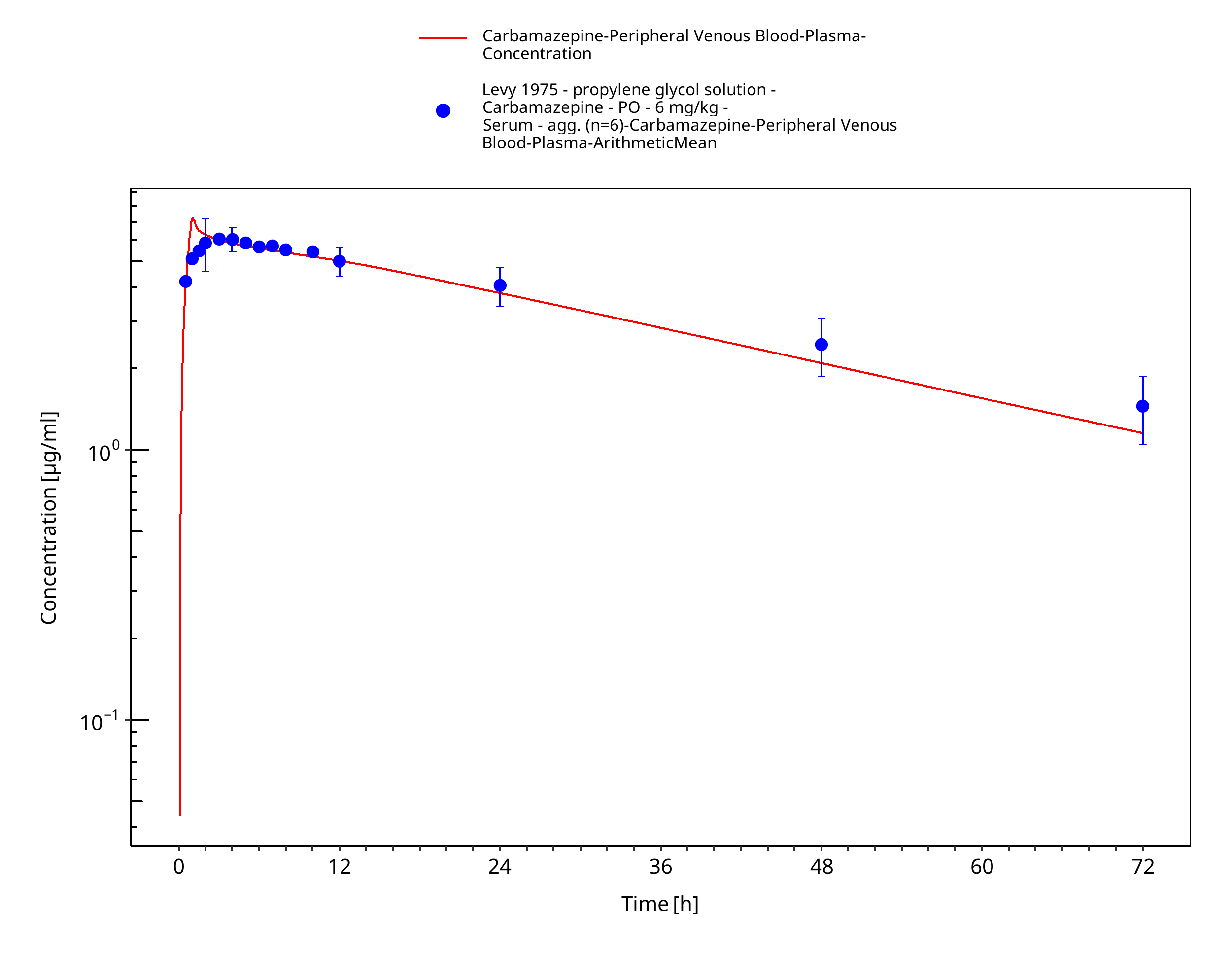
Figure 3-32: Levy1975_6mg-kg_sd_solution_fasted
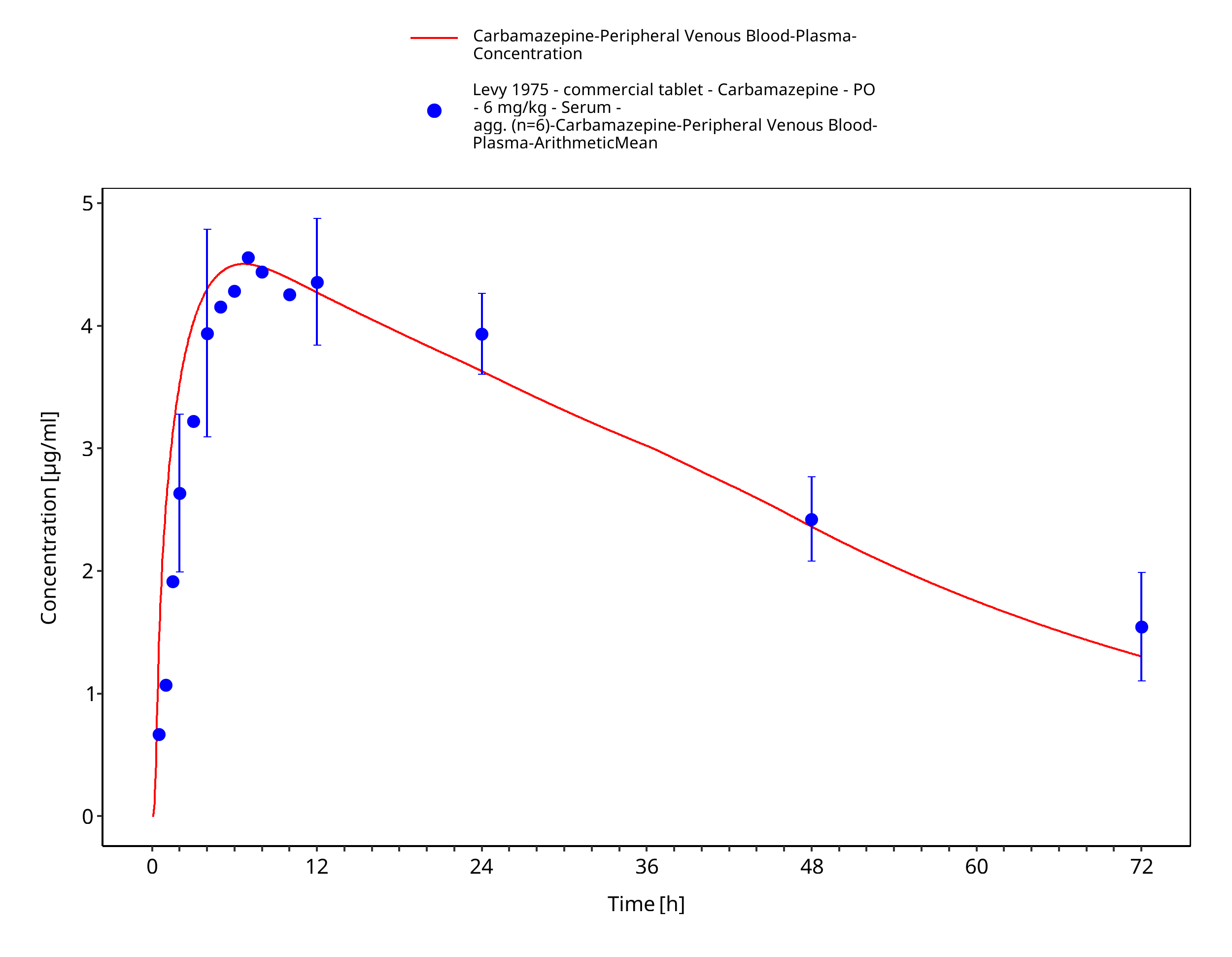
Figure 3-33: Levy1975_6mg-kg_sd_tabIR_fasted
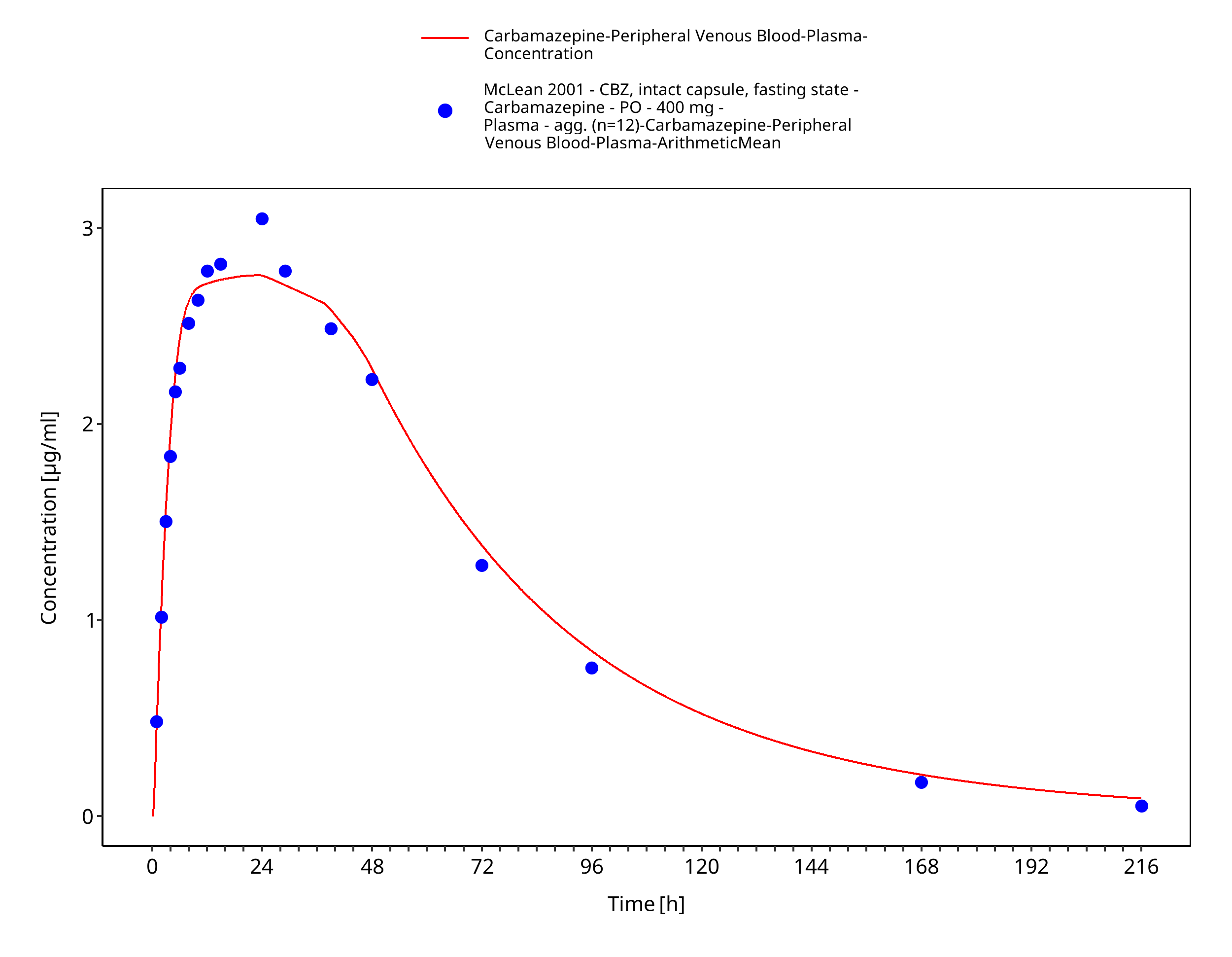
Figure 3-34: McLean2001_400mg_sd_capXR_fasted
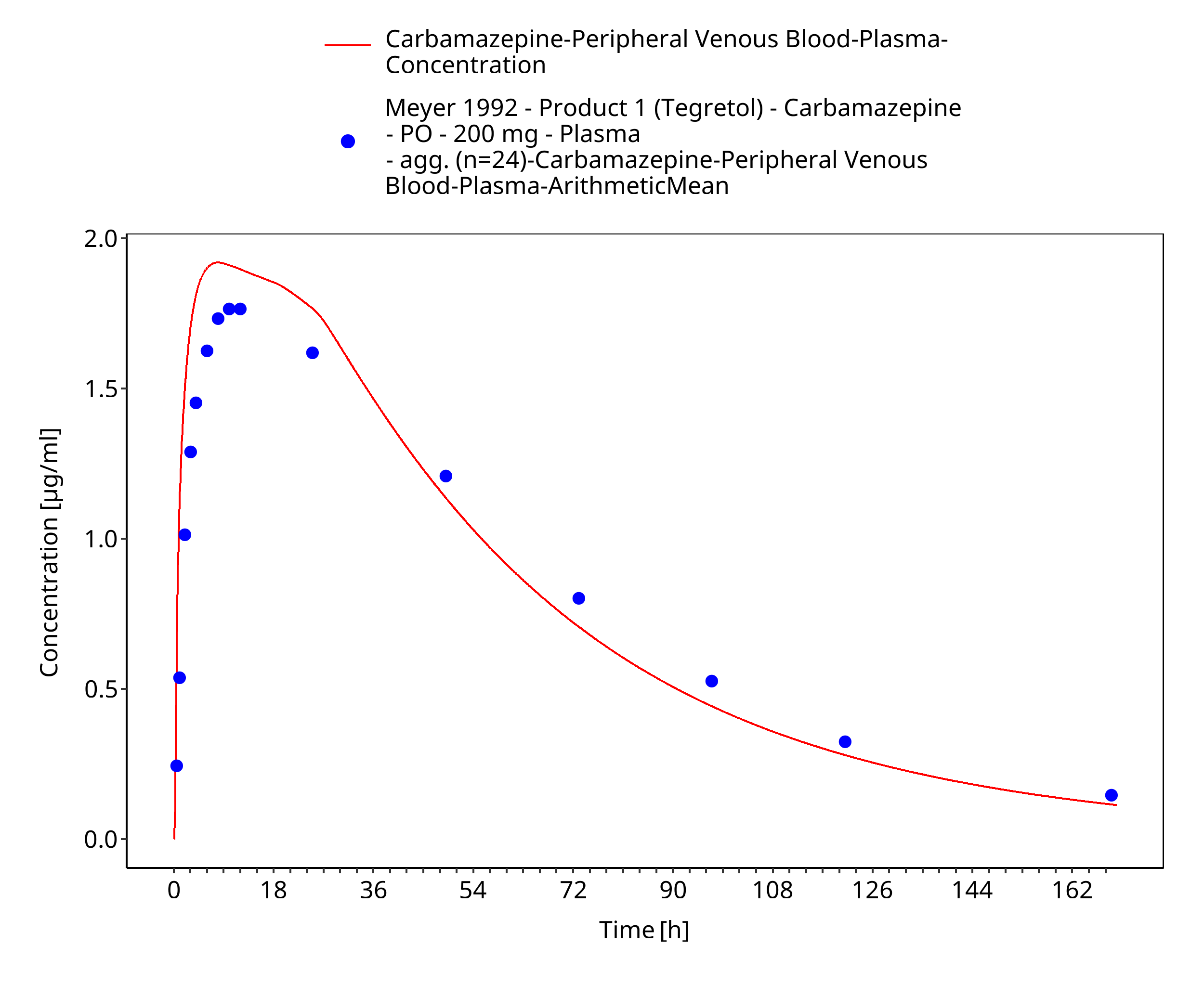
Figure 3-35: Time Profile Analysis
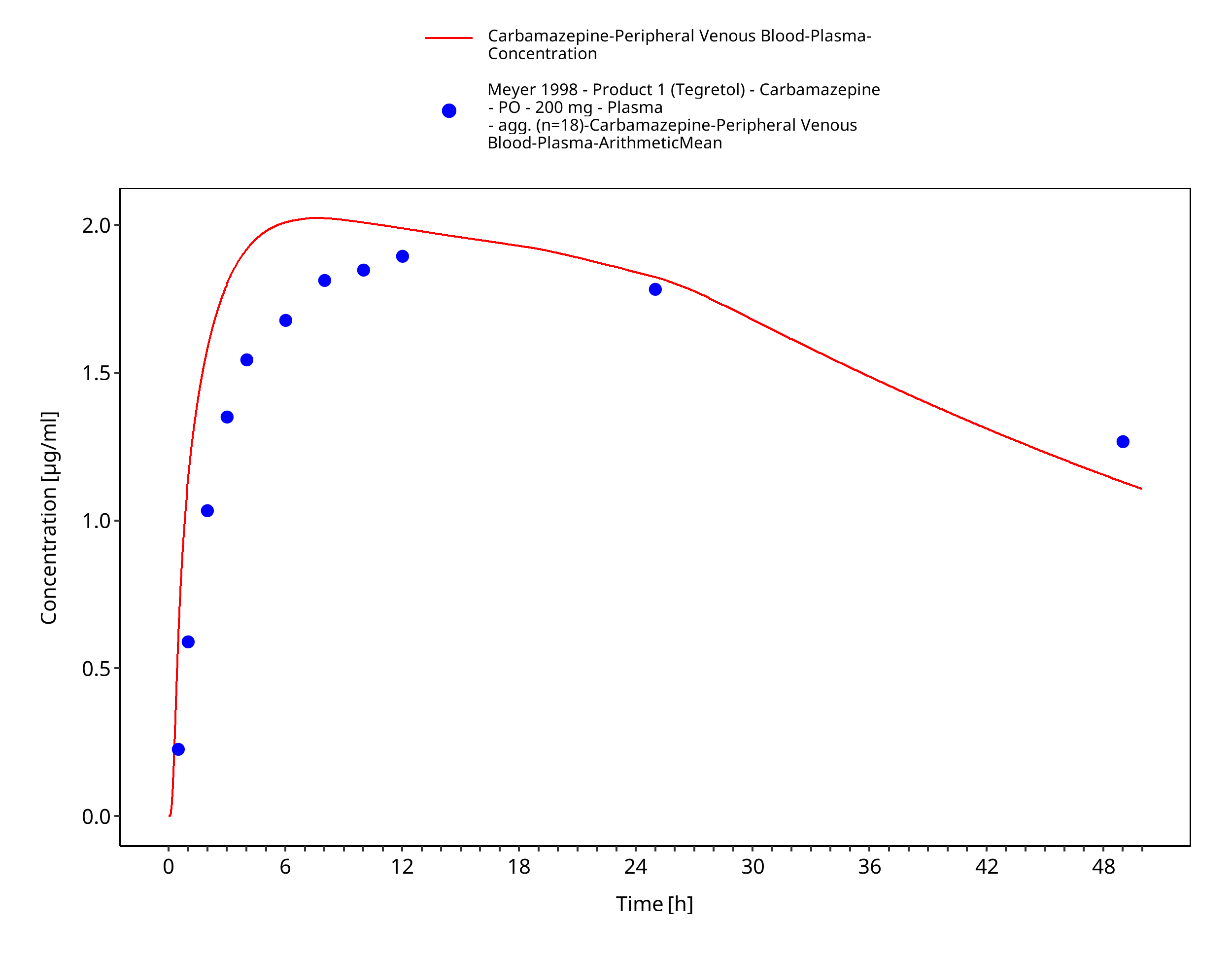
Figure 3-36: Time Profile Analysis
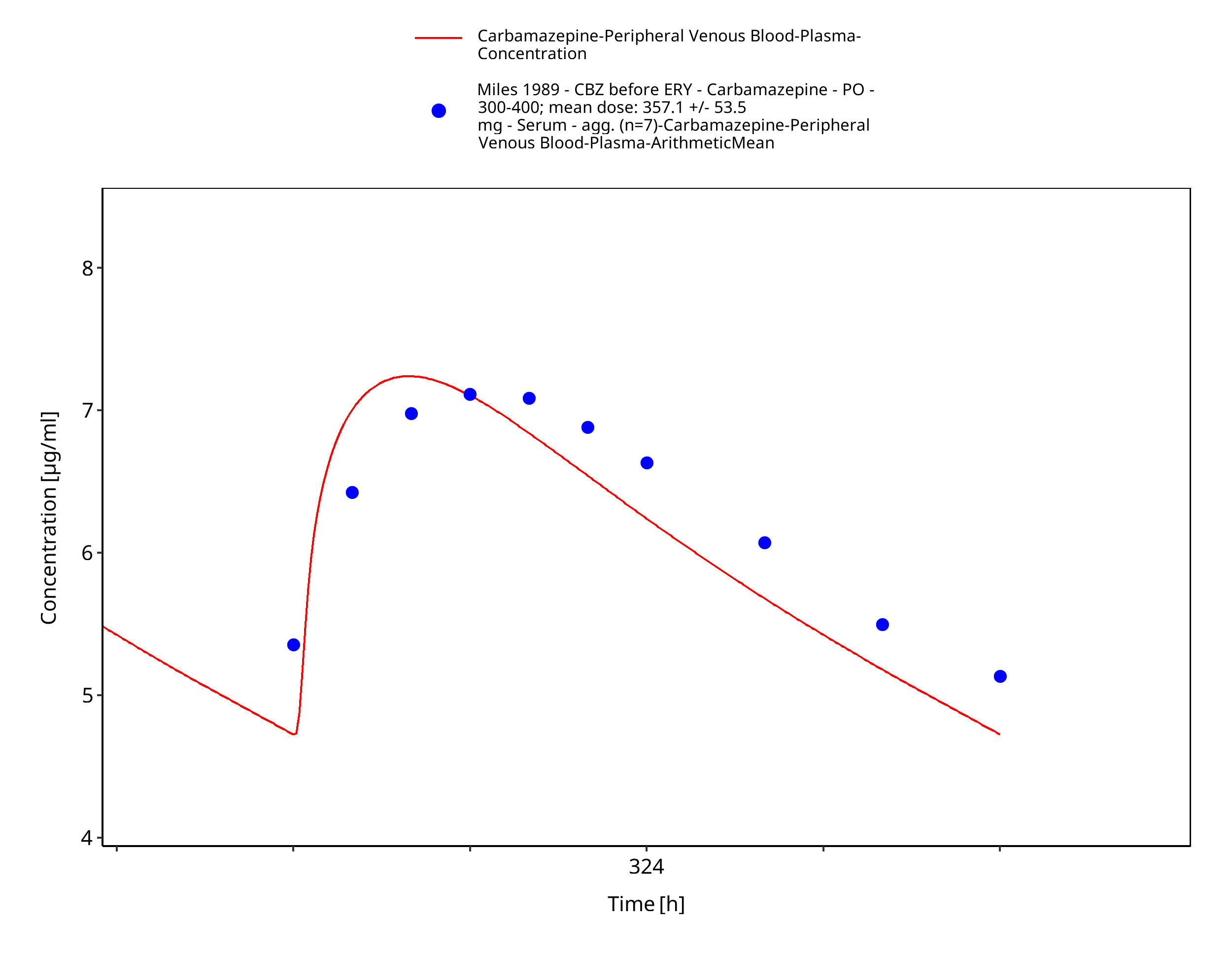
Figure 3-37: Miles1989_control_357_md_tabIR
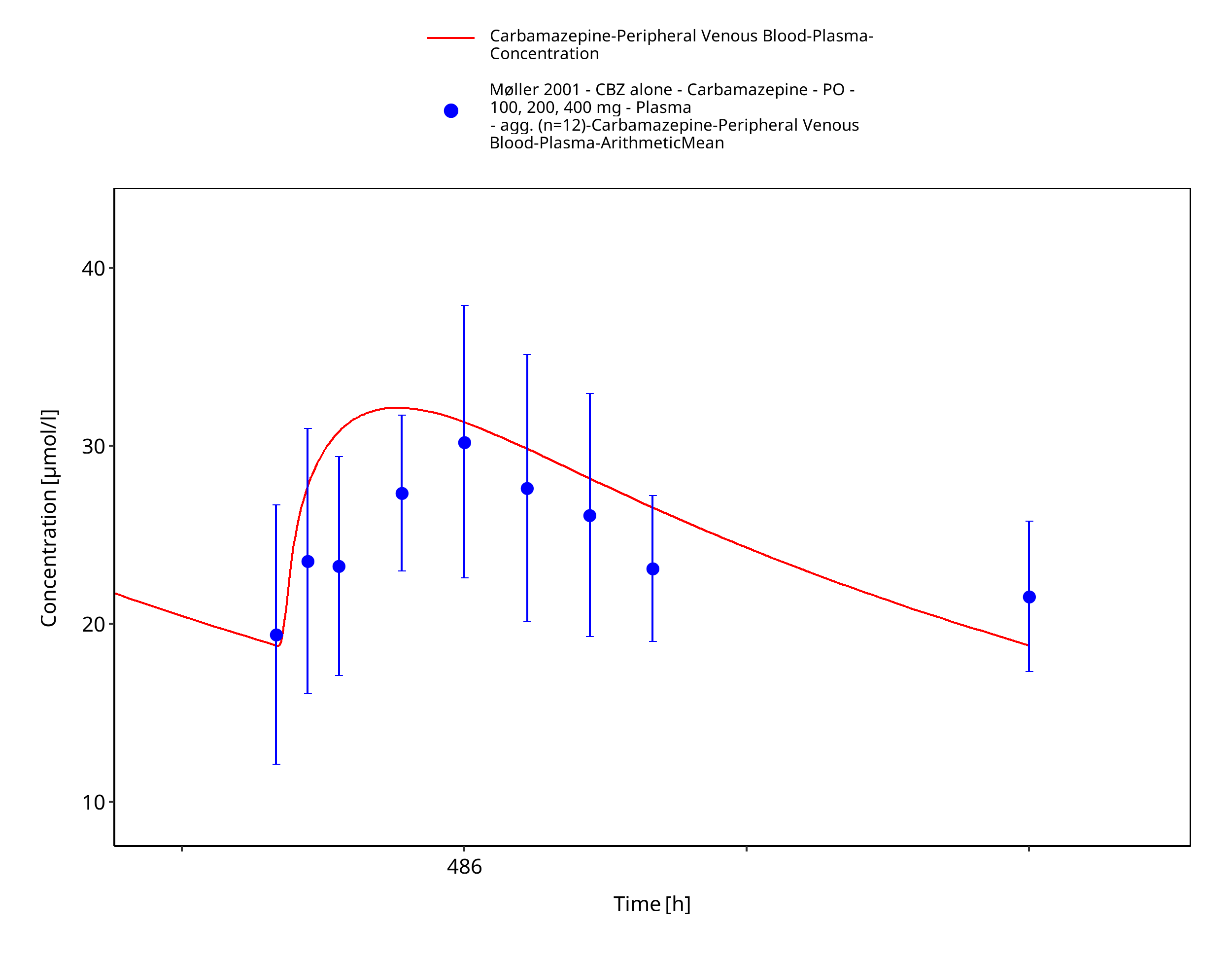
Figure 3-38: Moller2001_100-200-400mg_md_tabIR
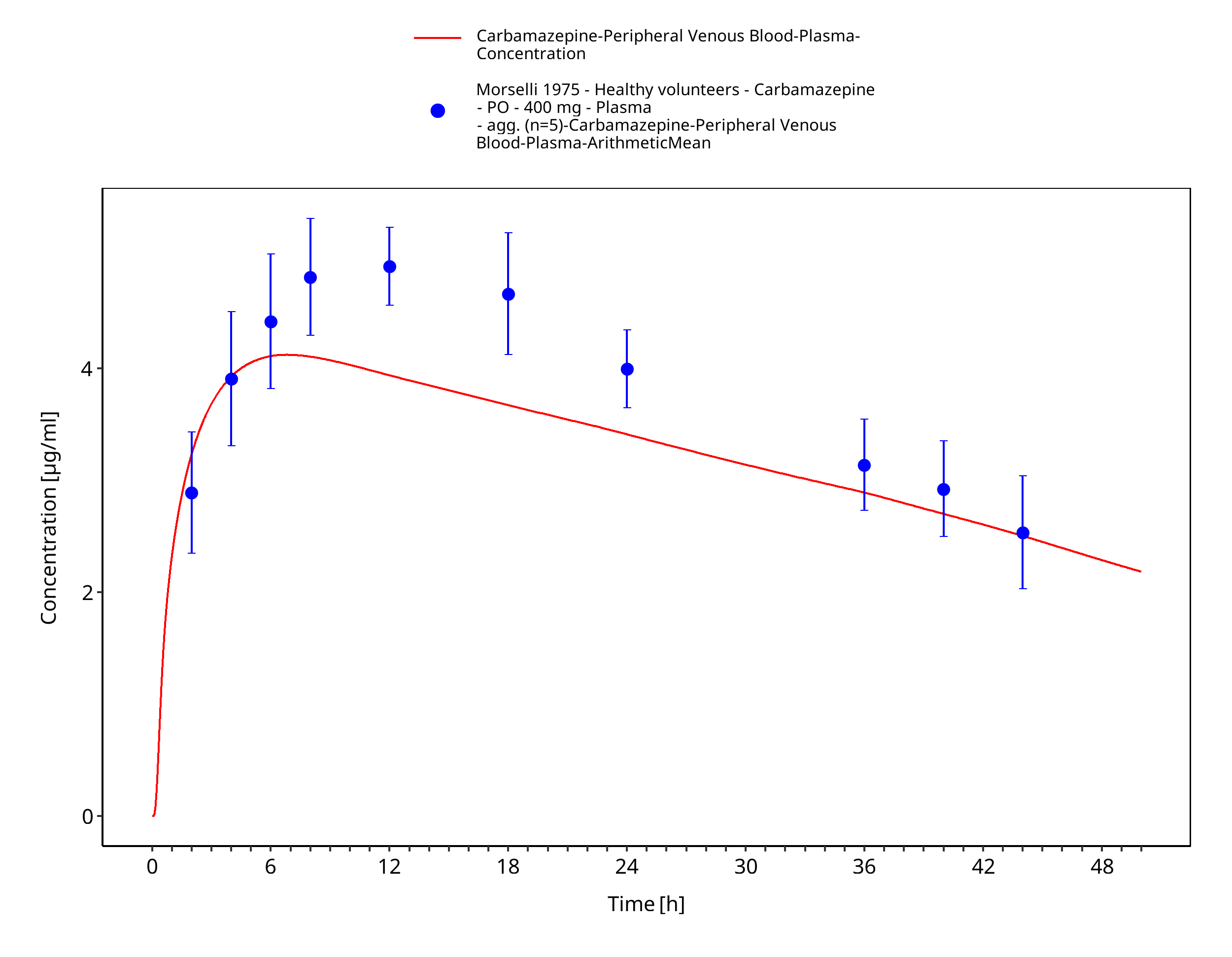
Figure 3-39: Morselli1975_healthy_400mg_sd_tabIR_plasma
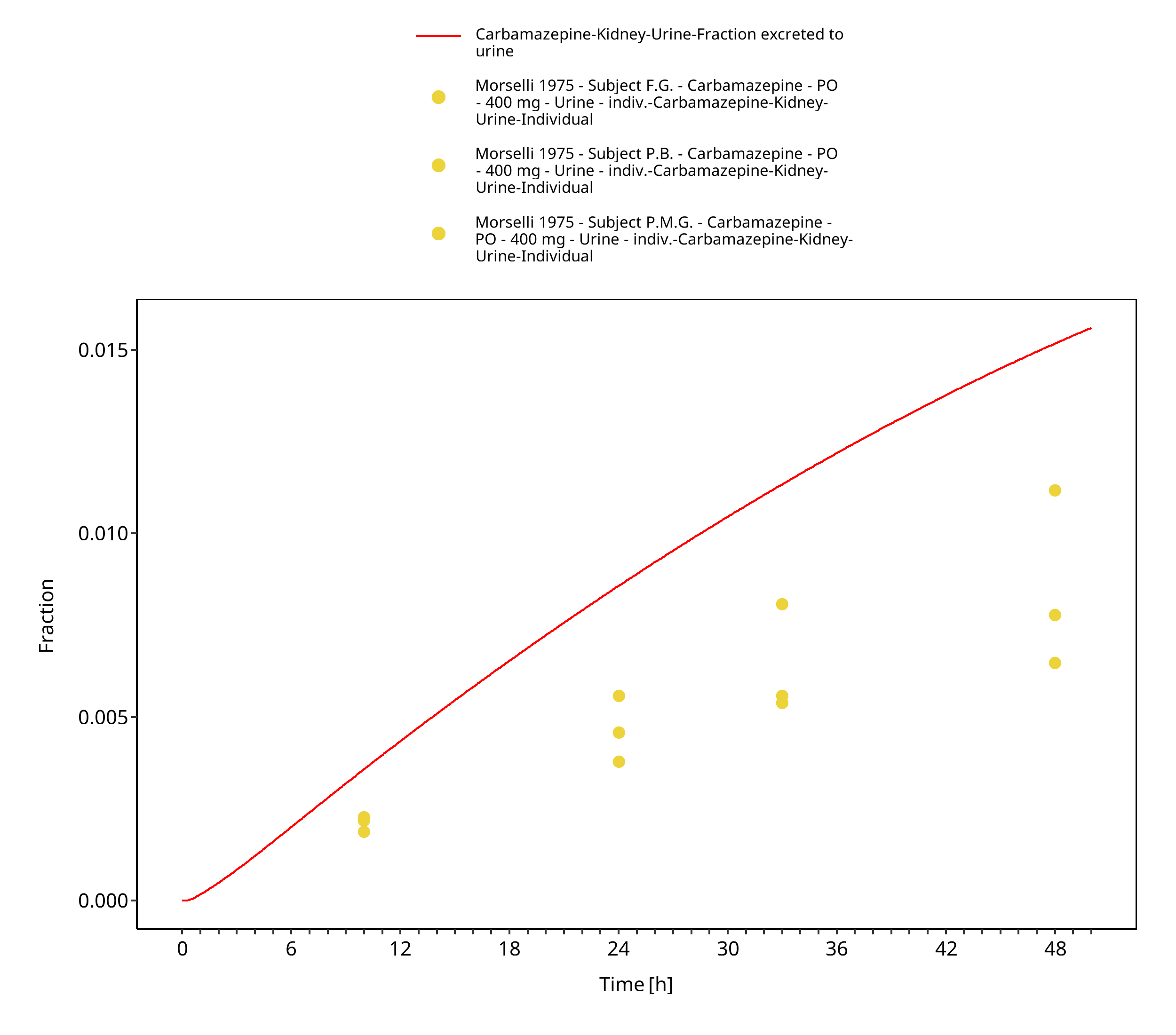
Figure 3-40: Morselli1975_healthy_400mg_sd_tabIR_urine
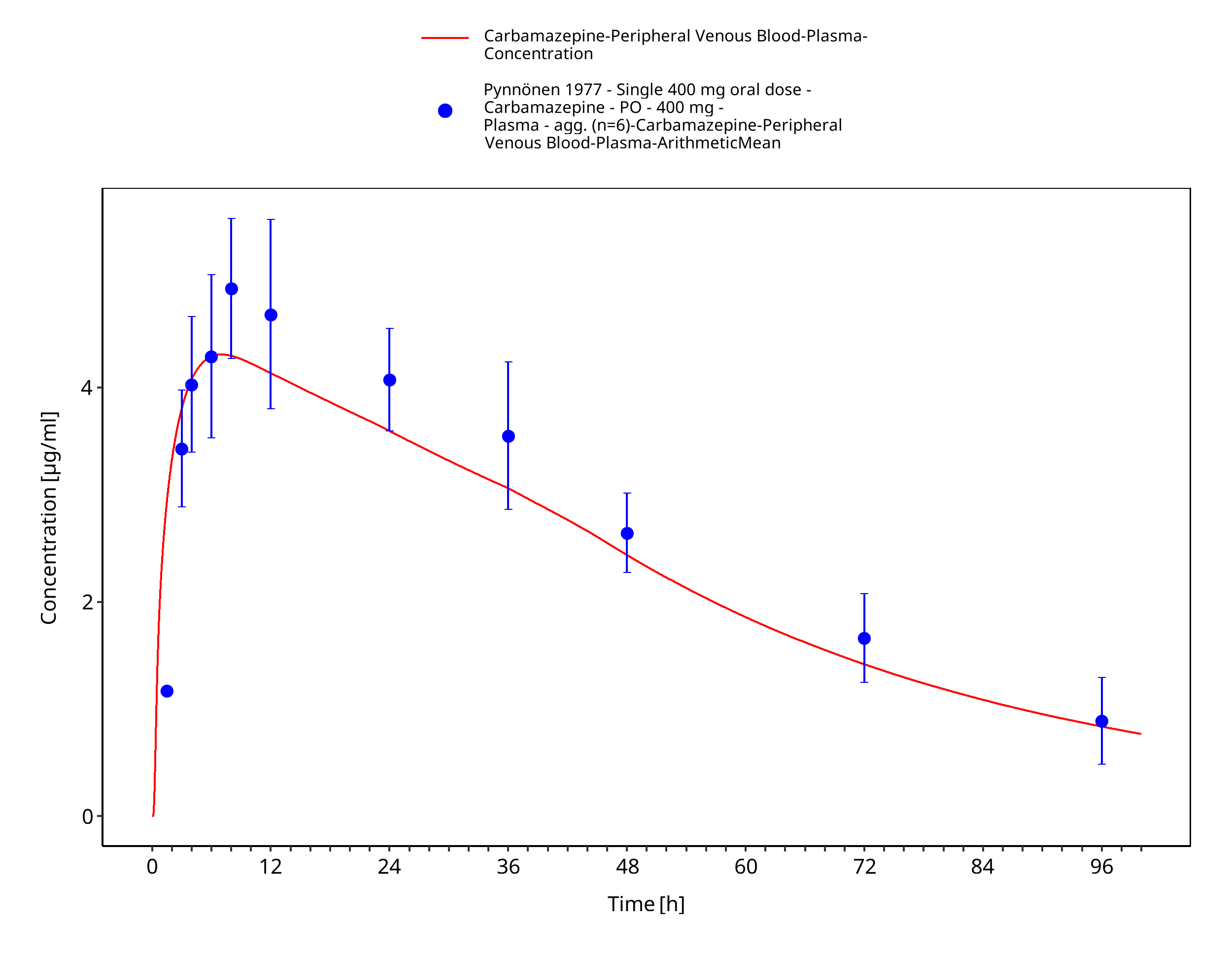
Figure 3-41: Pynnoenen1977_400mg_sd_tabIR_plasma
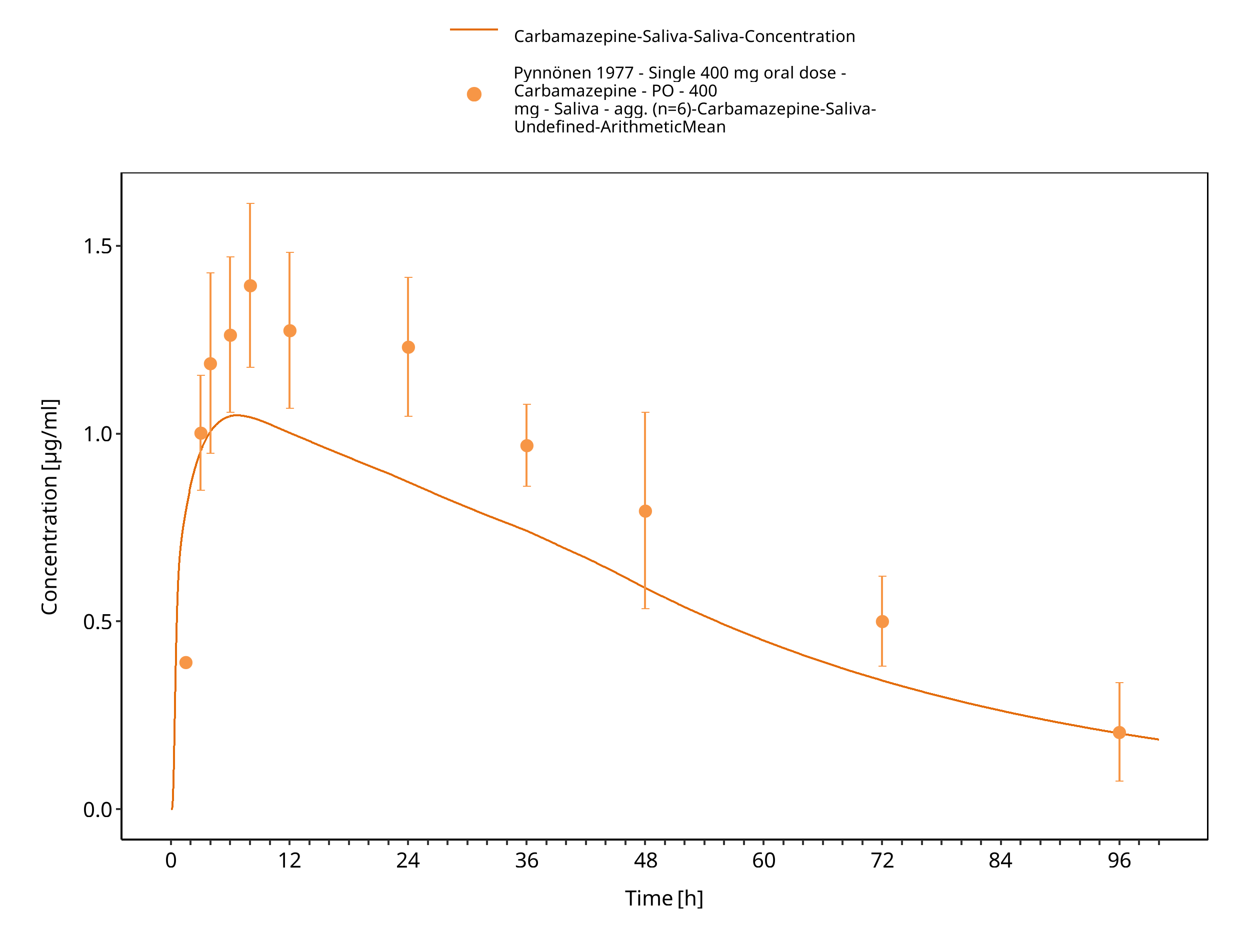
Figure 3-42: Pynnoenen1977_400mg_sd_tabIR_saliva
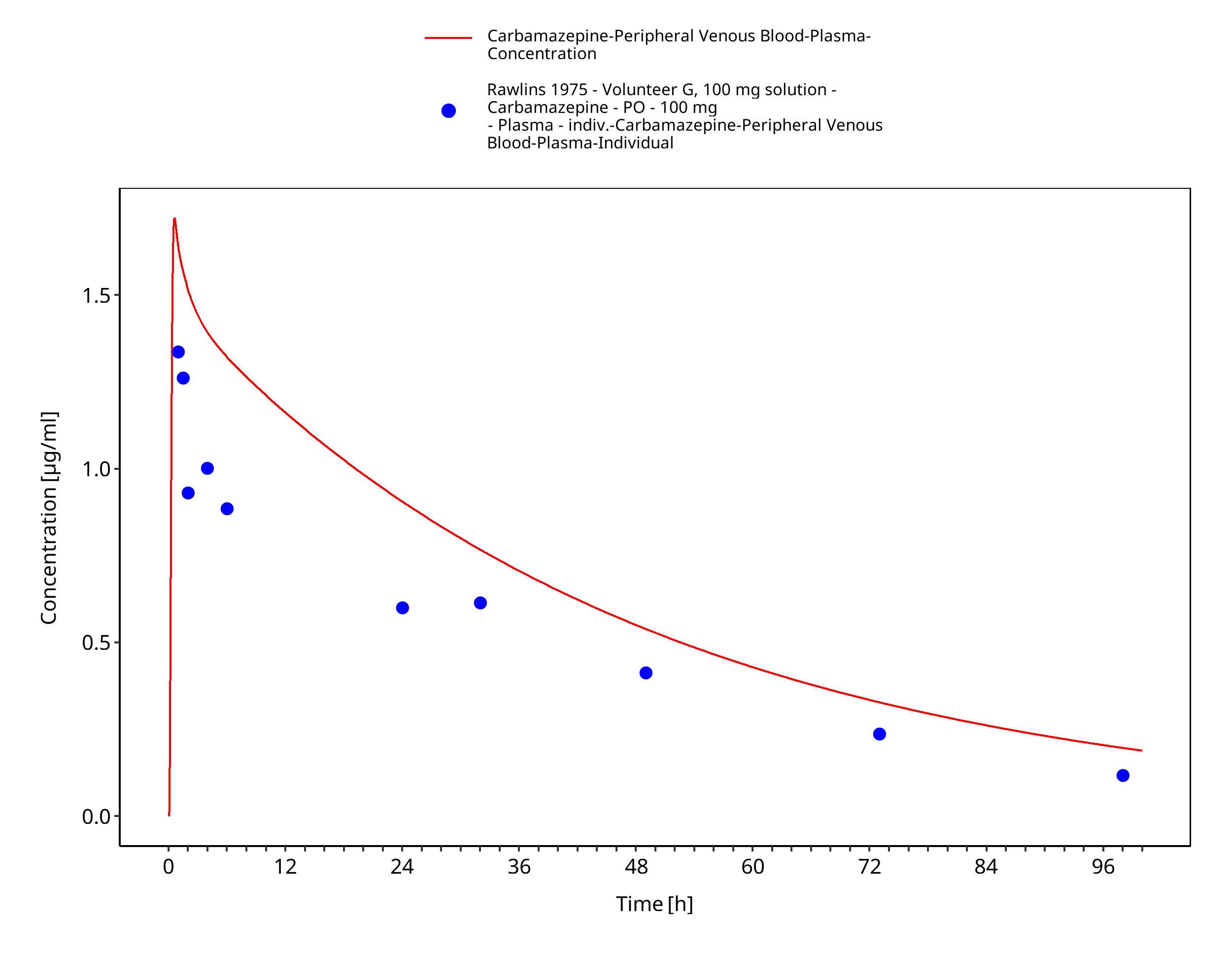
Figure 3-43: Rawlins1975_100mg_sd_sol
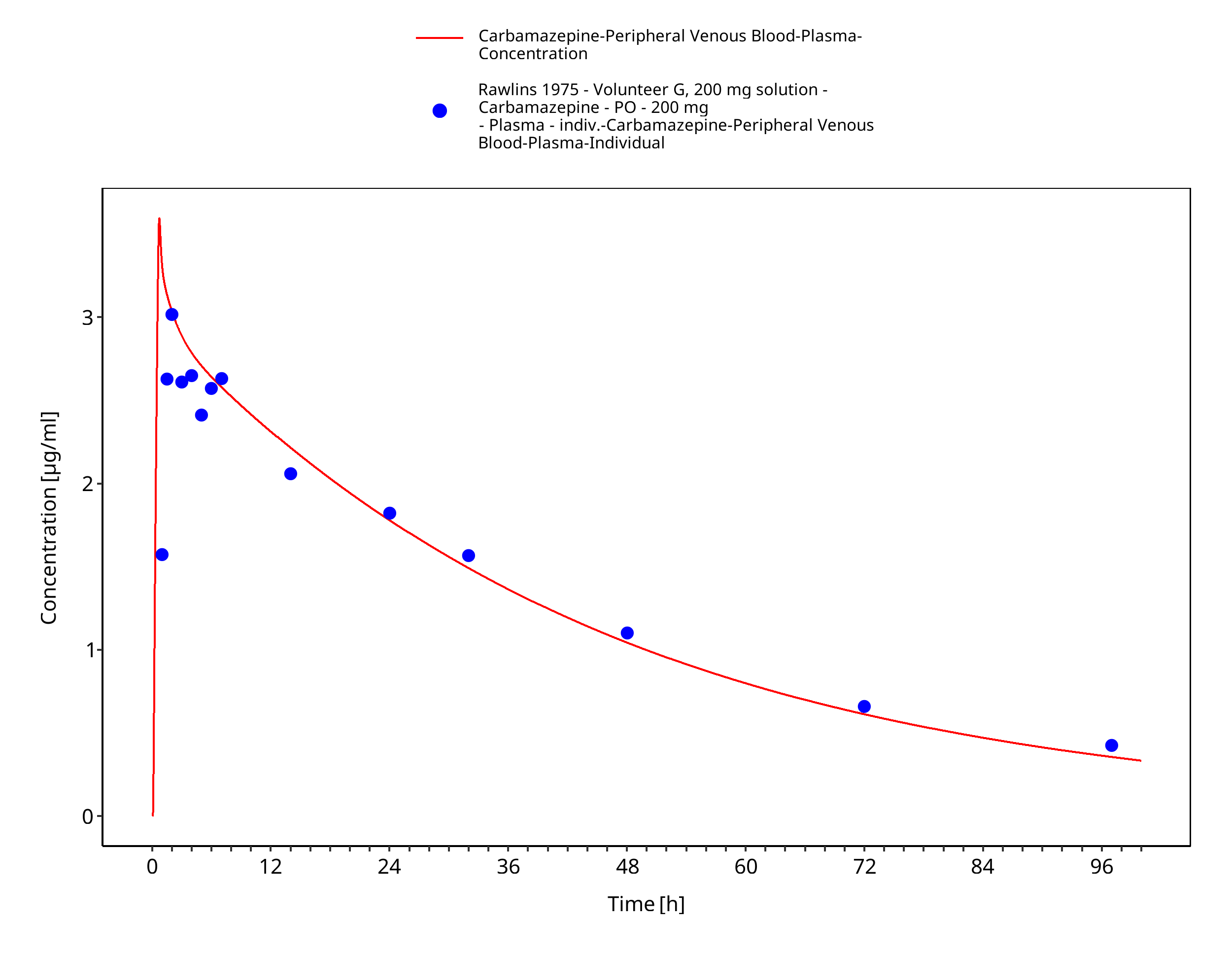
Figure 3-44: Rawlins1975_200mg_sd_sol
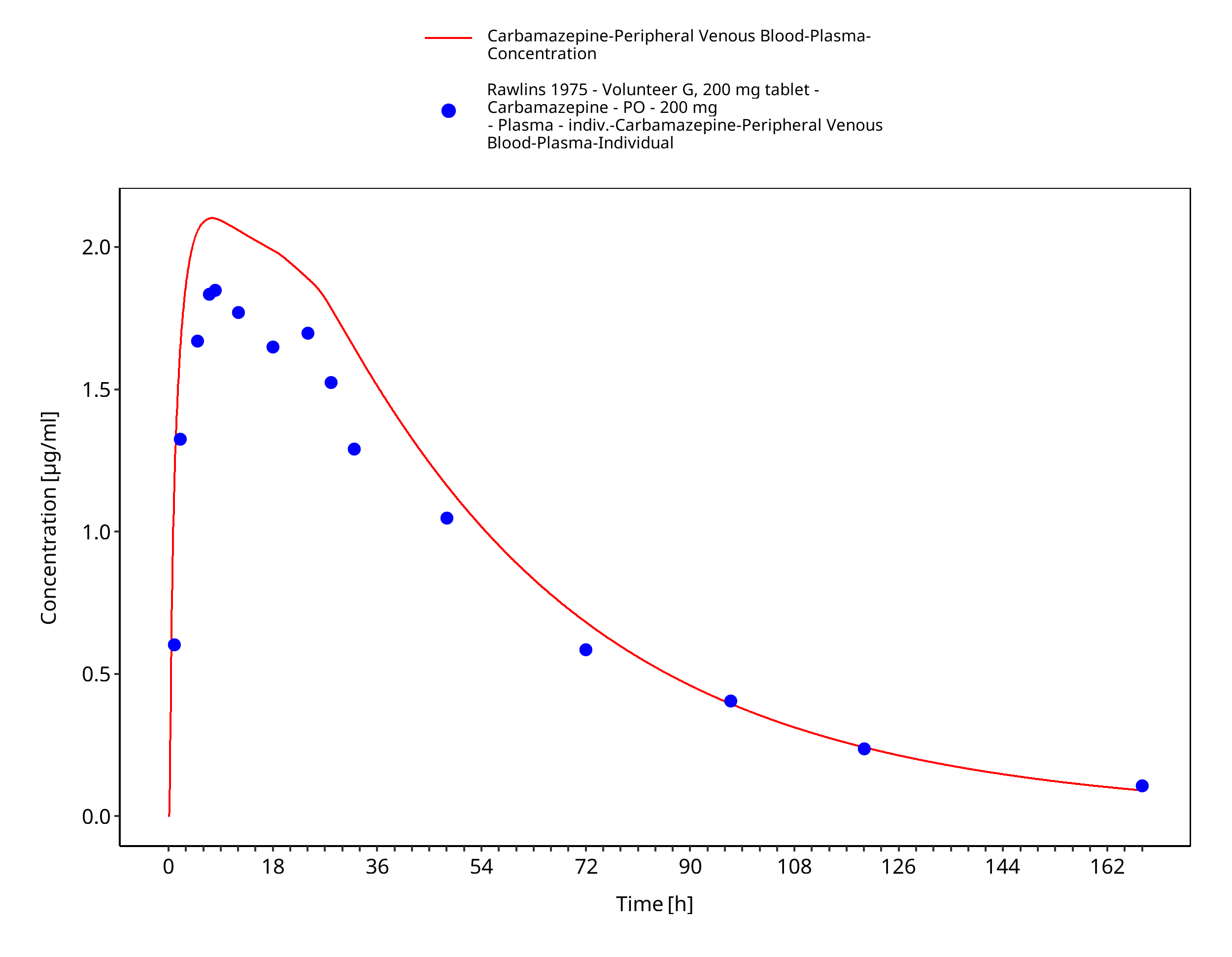
Figure 3-45: Rawlins1975_200mg_sd_sol
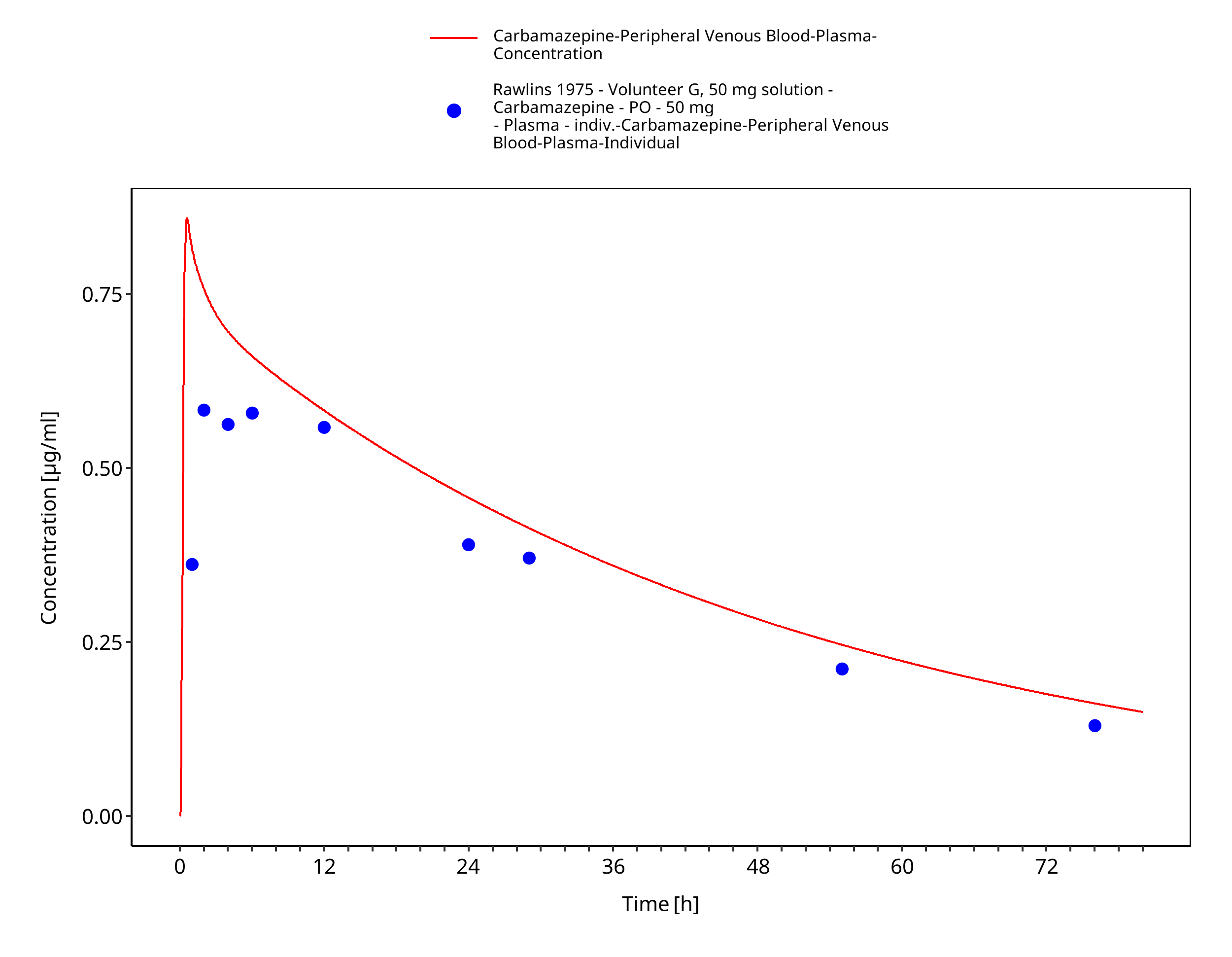
Figure 3-46: Rawlins1975_50mg_sd_sol
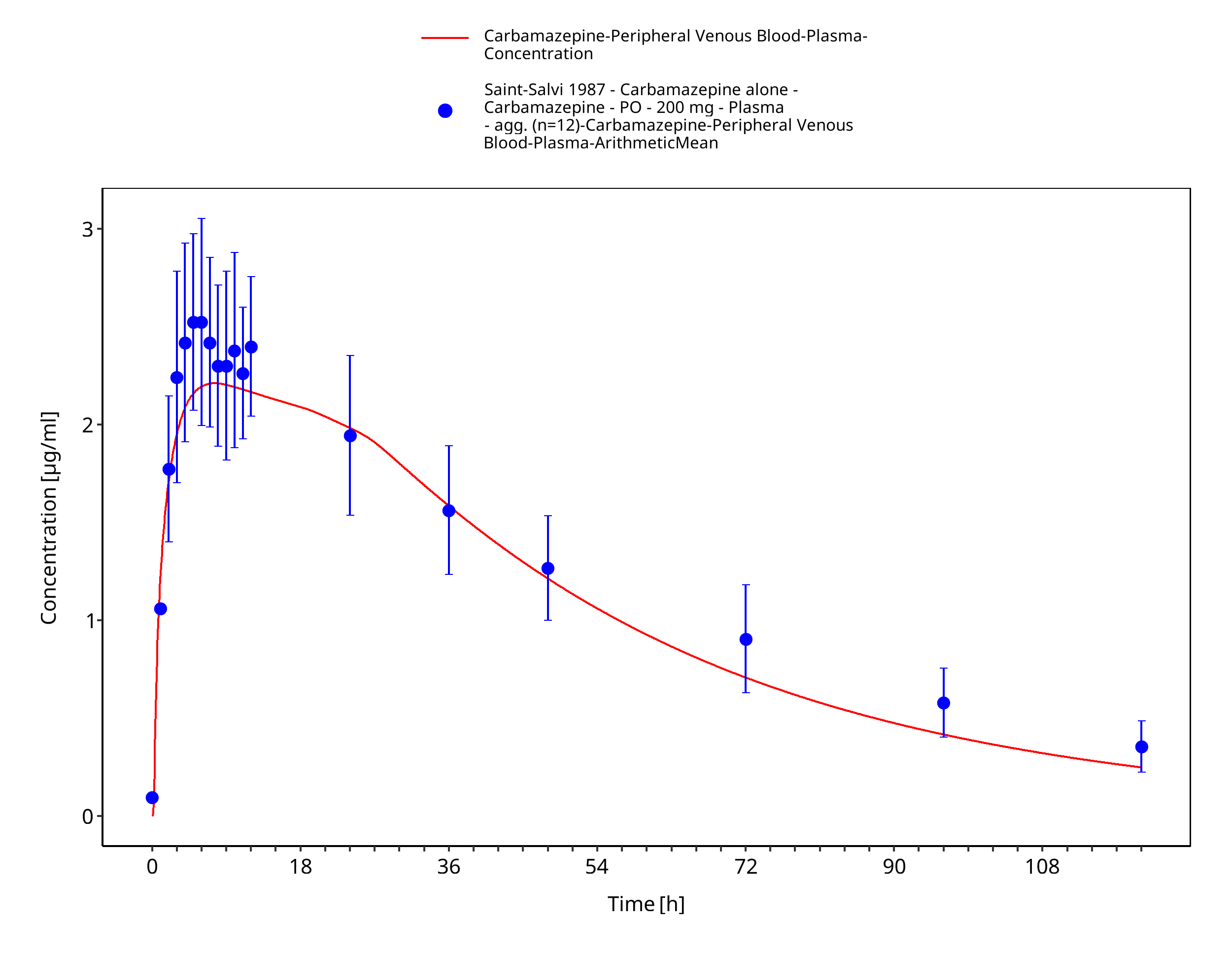
Figure 3-47: SaintSalvi1987_200mg_sd_tabIR
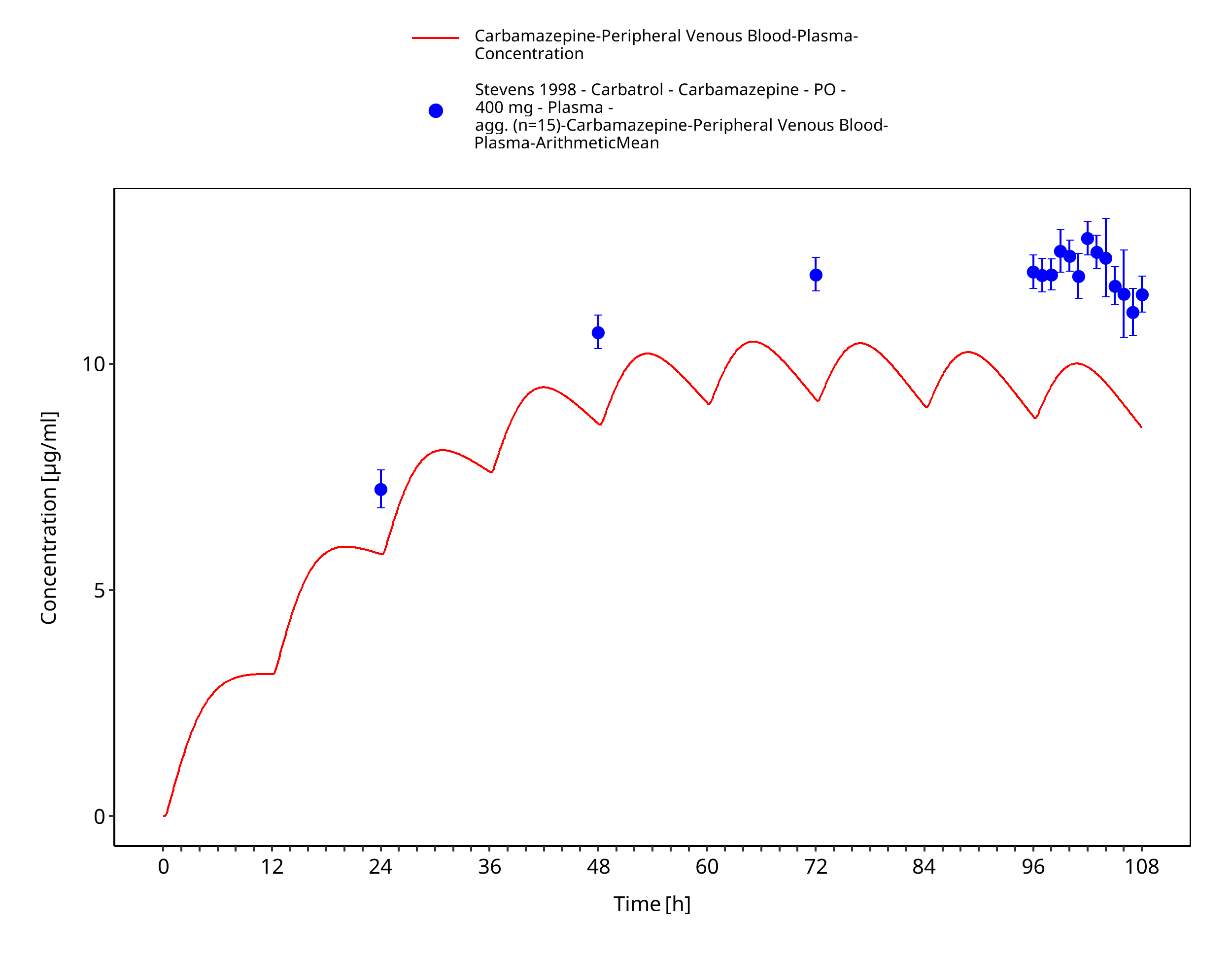
Figure 3-48: Stevens1998_400mg_bid_CarbatrolXR
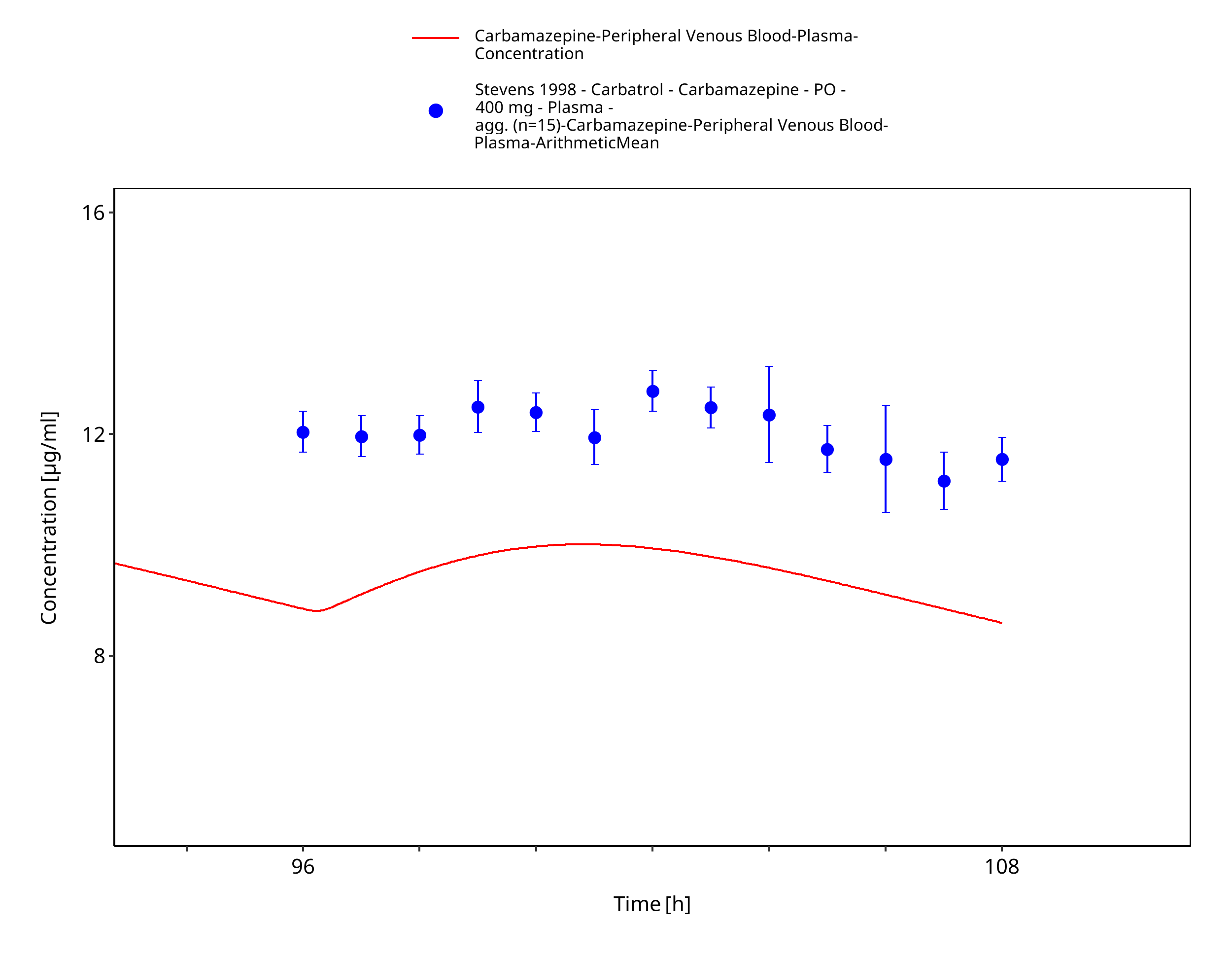
Figure 3-49: Stevens1998_400mg_bid_CarbatrolXR - last dose
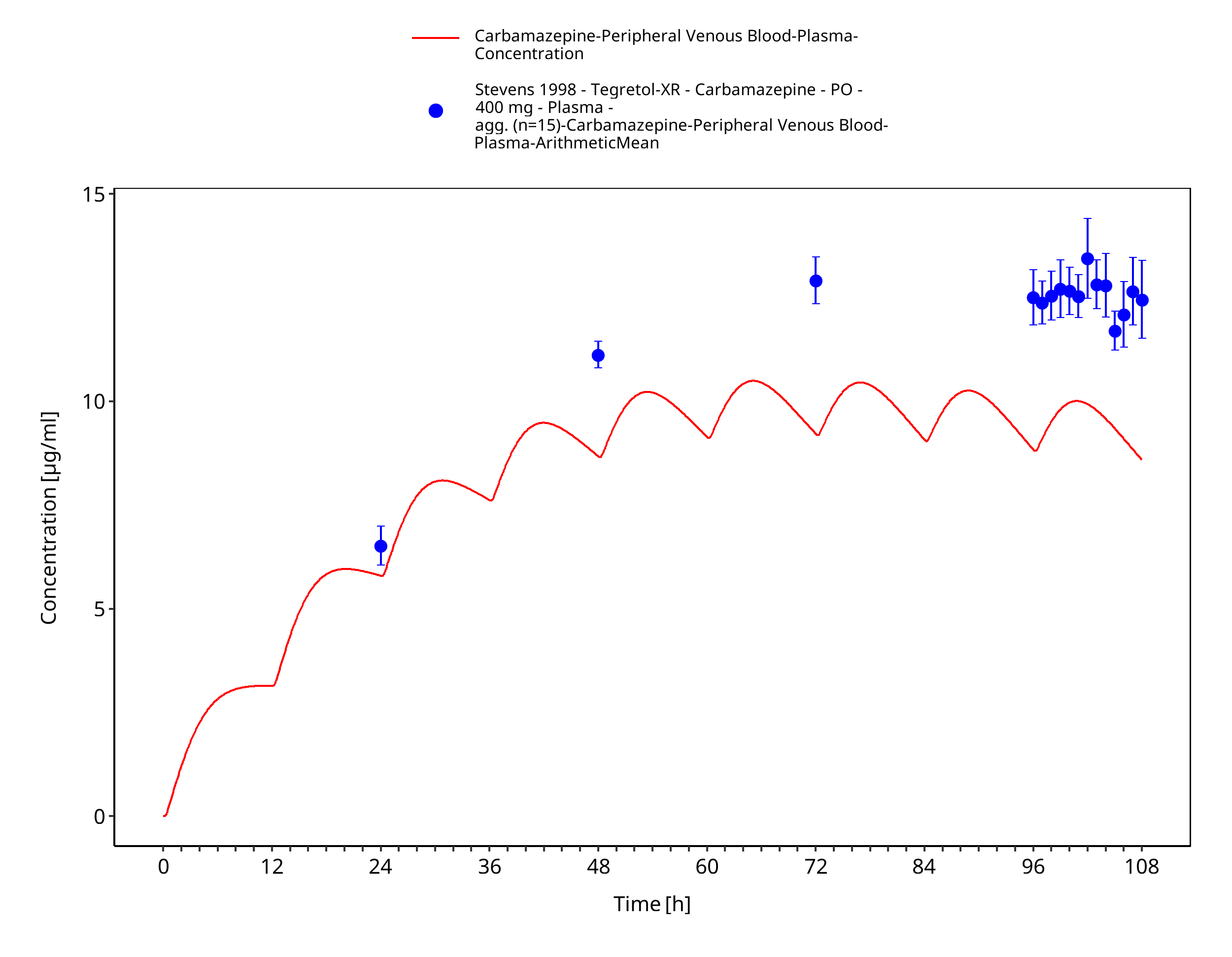
Figure 3-50: Stevens1998_400mg_bid_TegretolXR
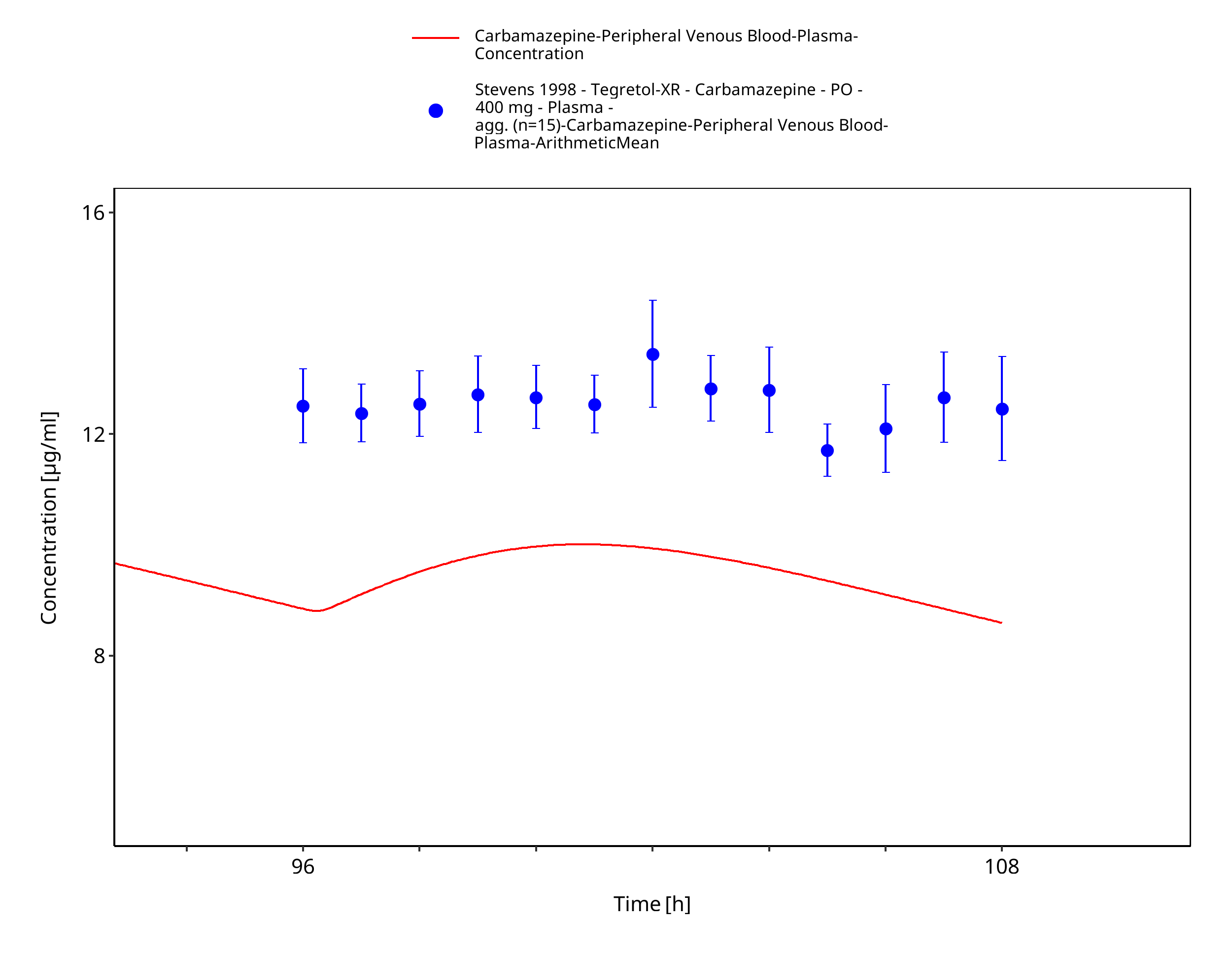
Figure 3-51: Stevens1998_400mg_bid_TegretolXR - last dose
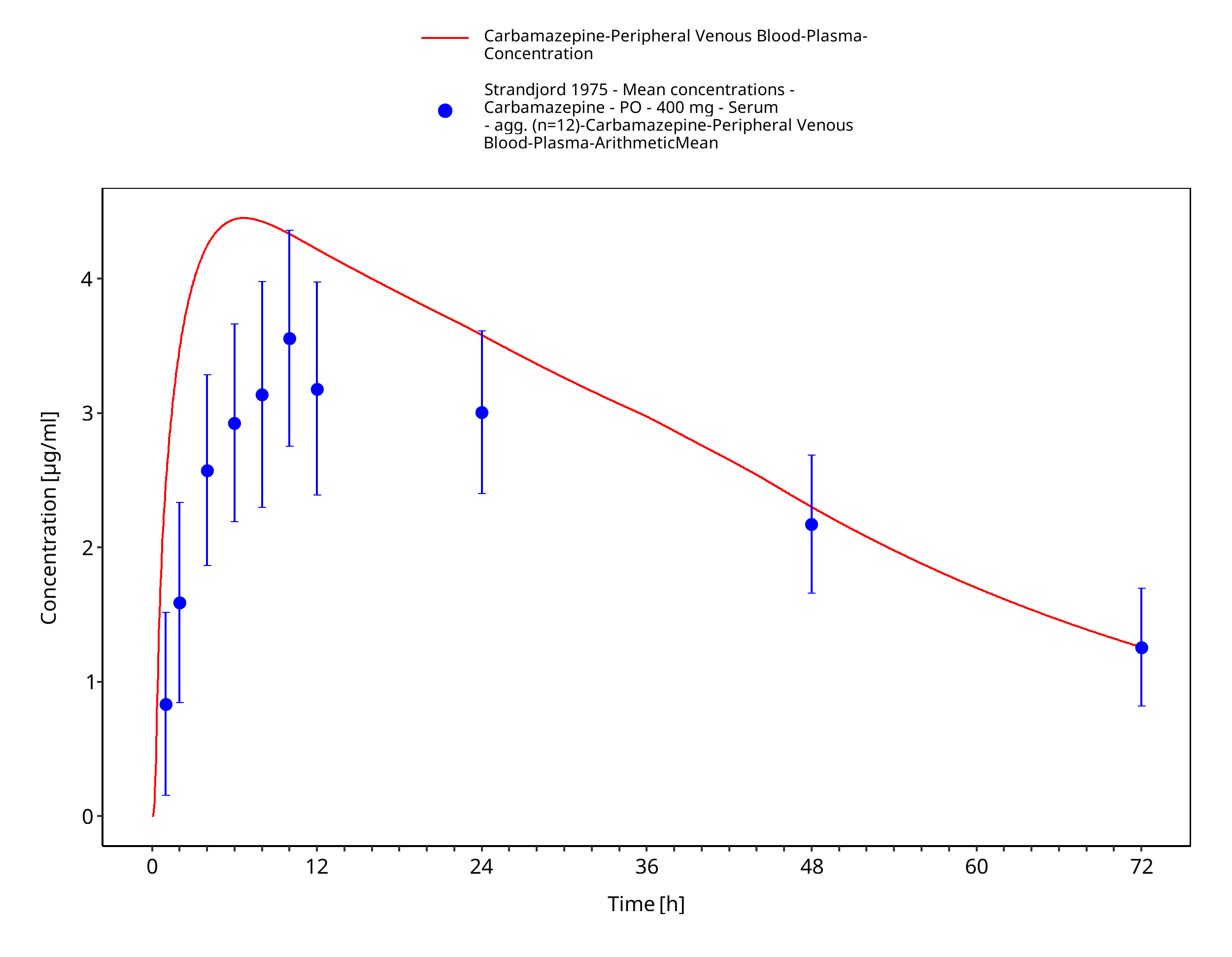
Figure 3-52: Strandjord1975_mean_400mg_sd_tabIR - observed mean data
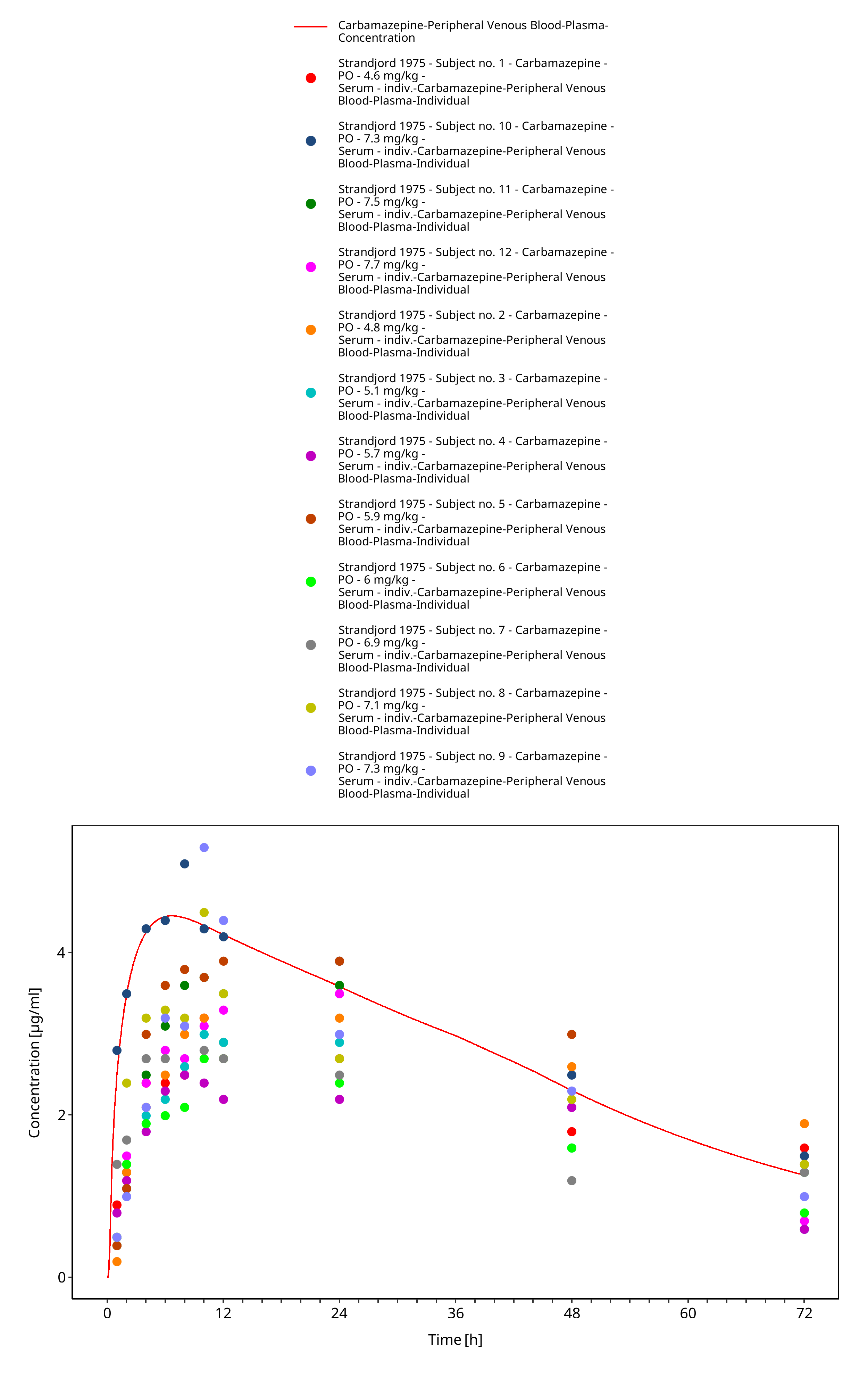
Figure 3-53: Strandjord1975_mean_400mg_sd_tabIR - observed individual data
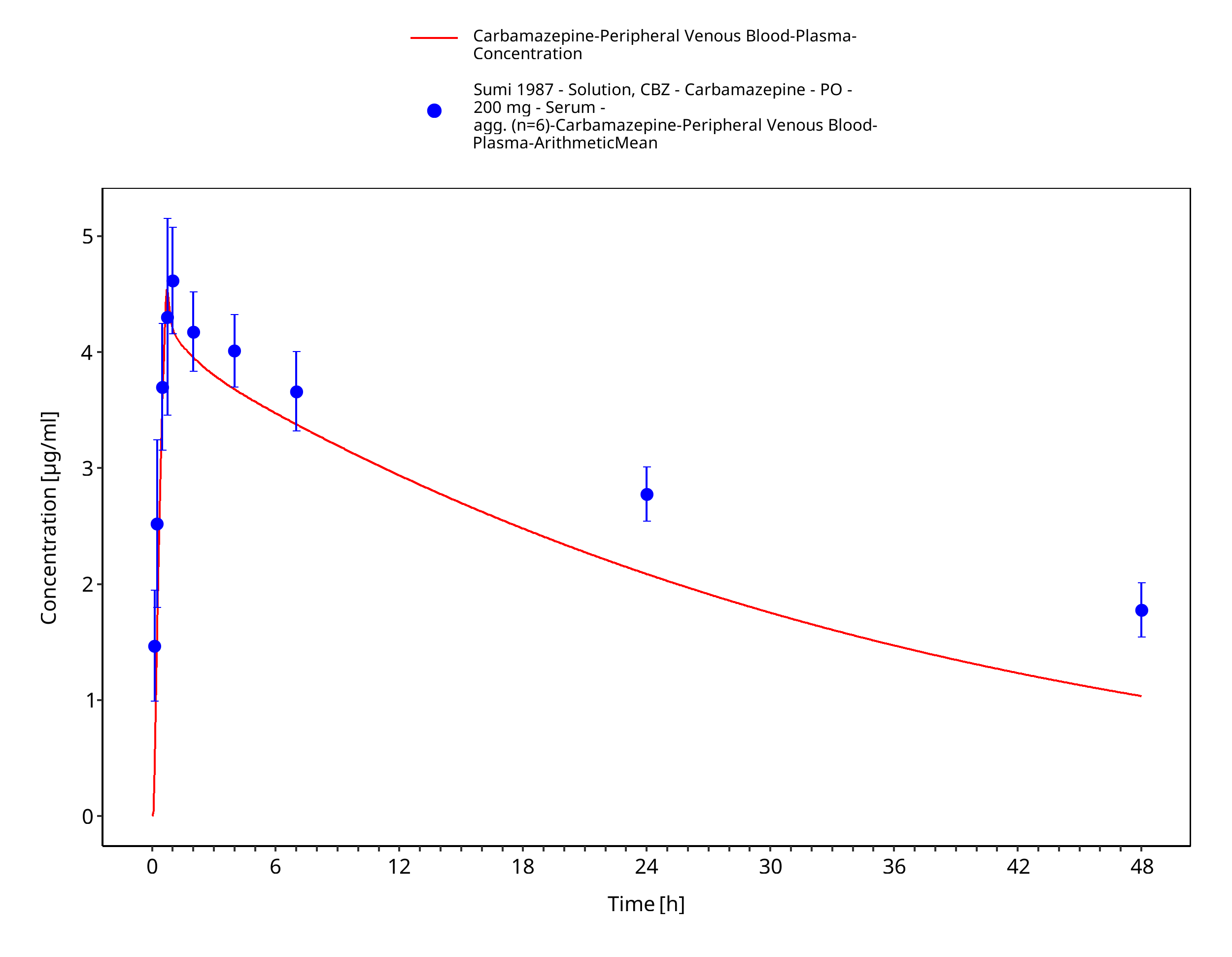
Figure 3-54: Sumi1987_200mg_sd_solution
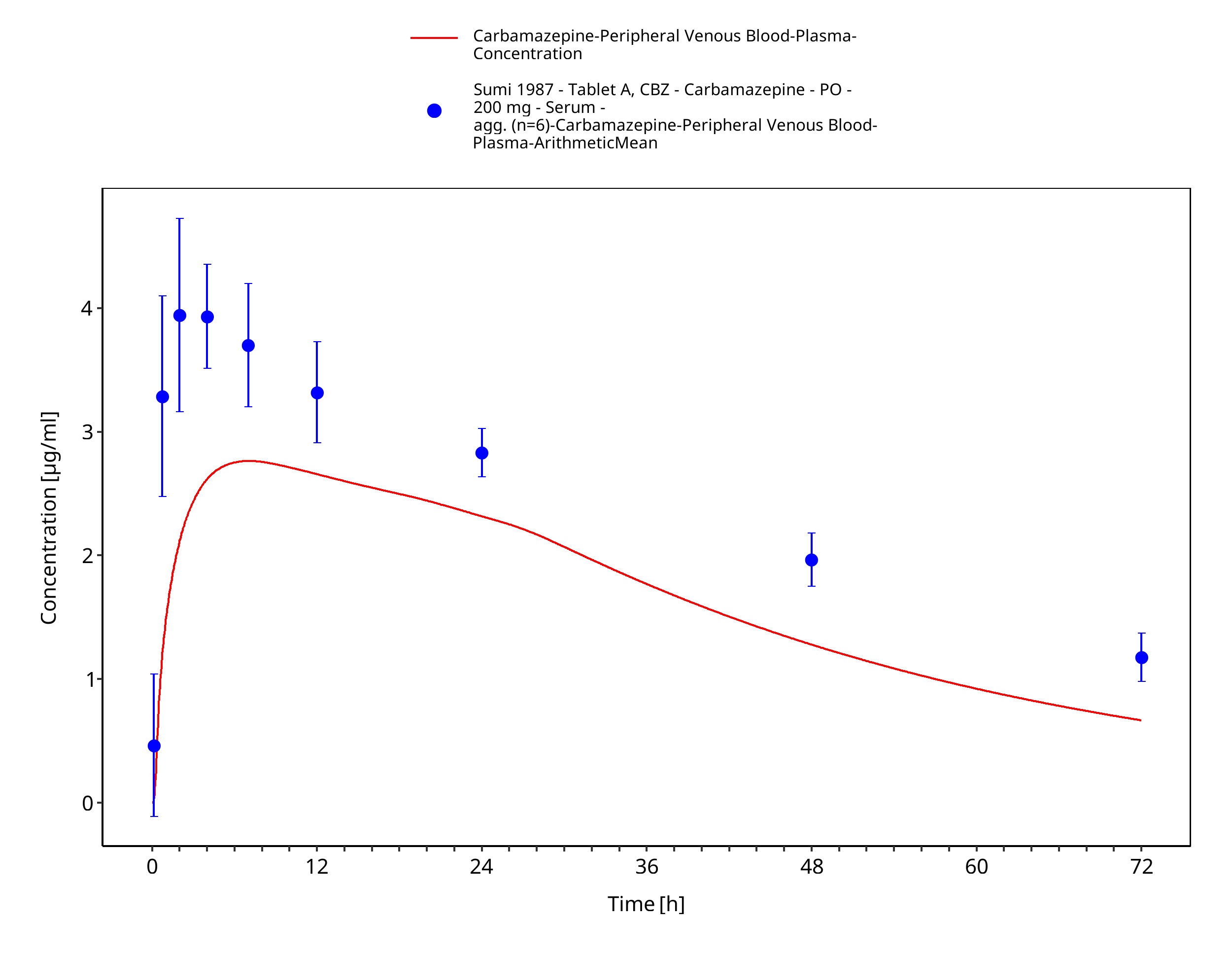
Figure 3-55: Sumi1987_200mg_sd_tabletA
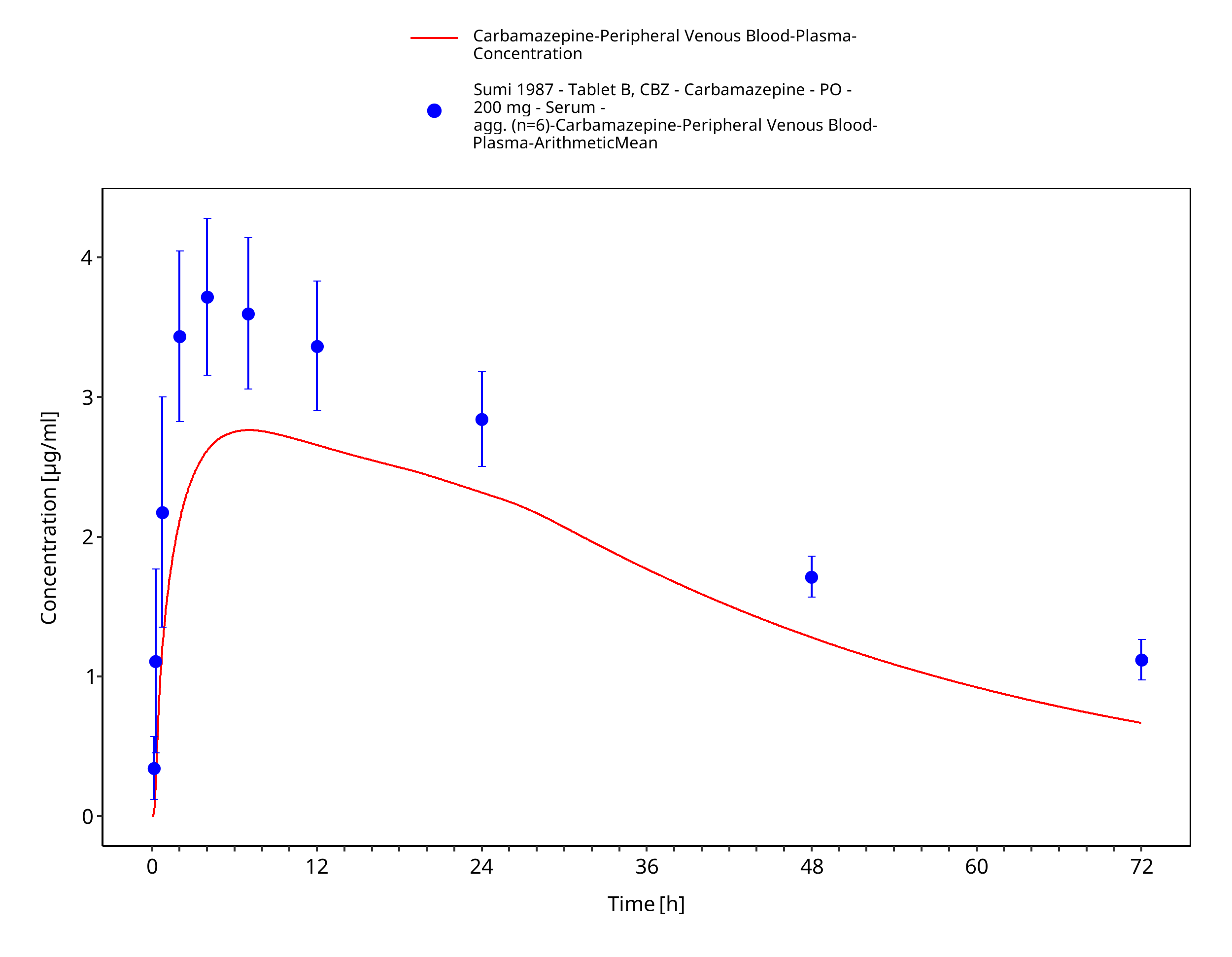
Figure 3-56: Sumi1987_200mg_sd_tabletB
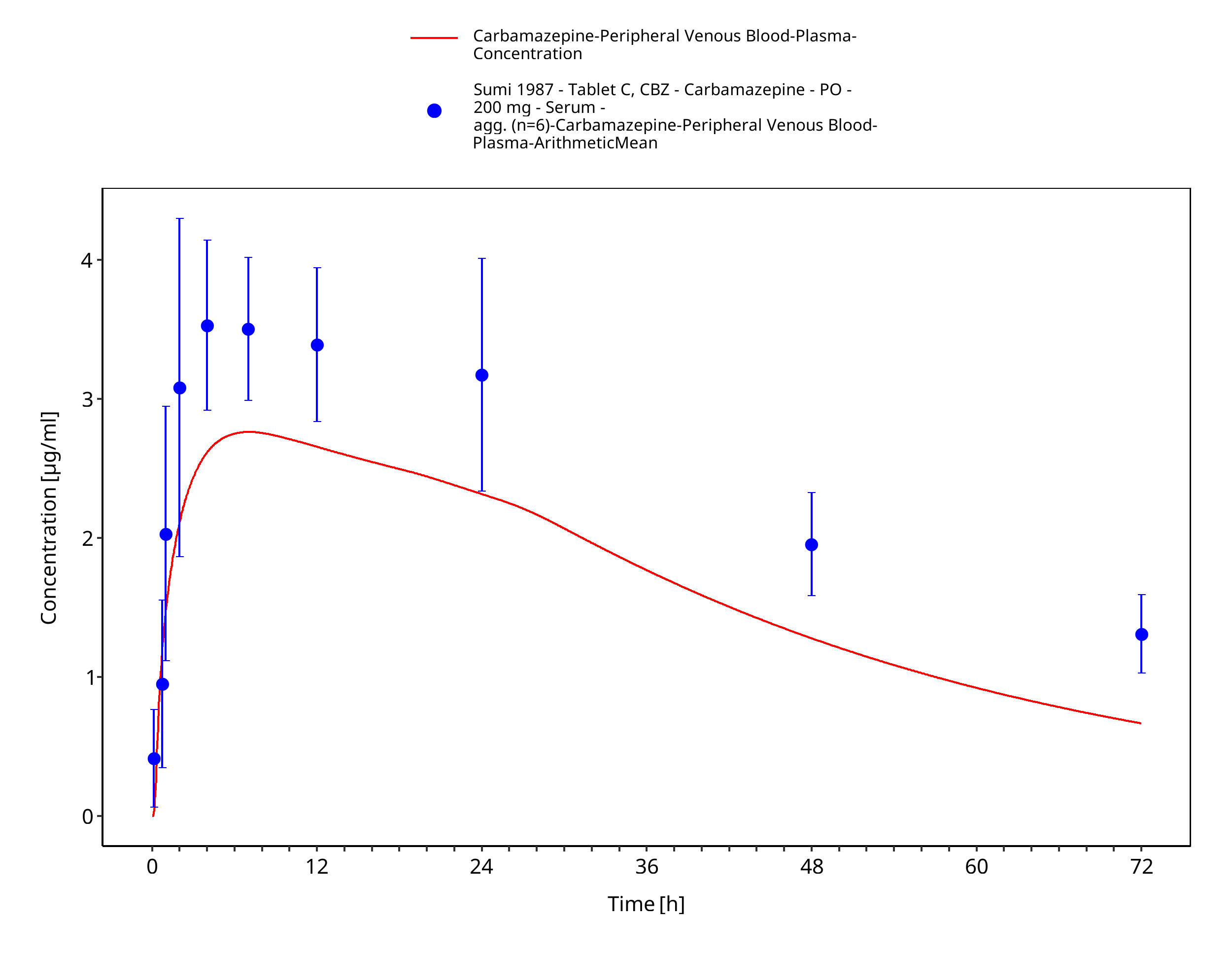
Figure 3-57: Sumi1987_200mg_sd_tabletC
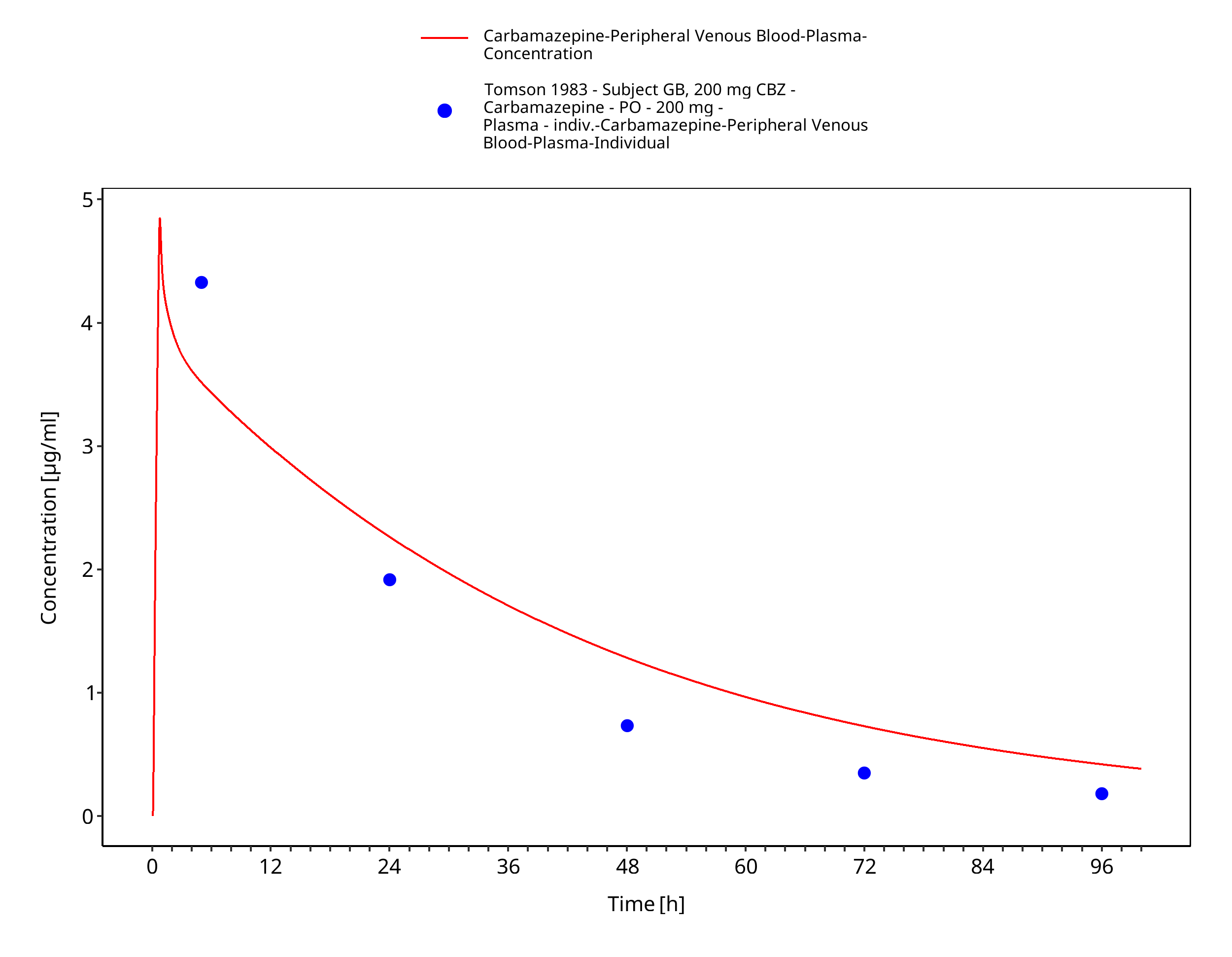
Figure 3-58: Tomson1983_GB_200mg_sd_susp
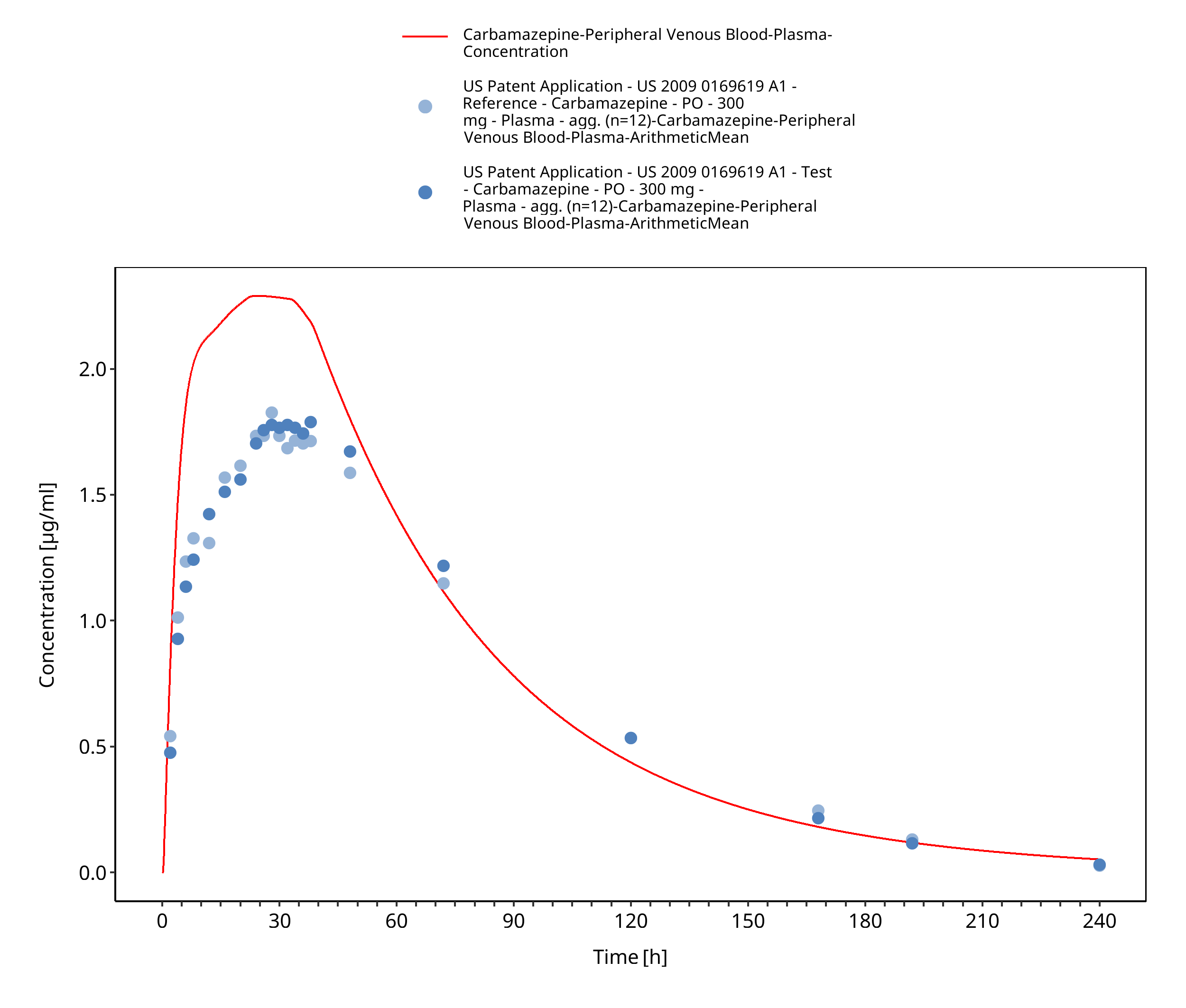
Figure 3-59: USPatent2009_300mg_sd_capXR
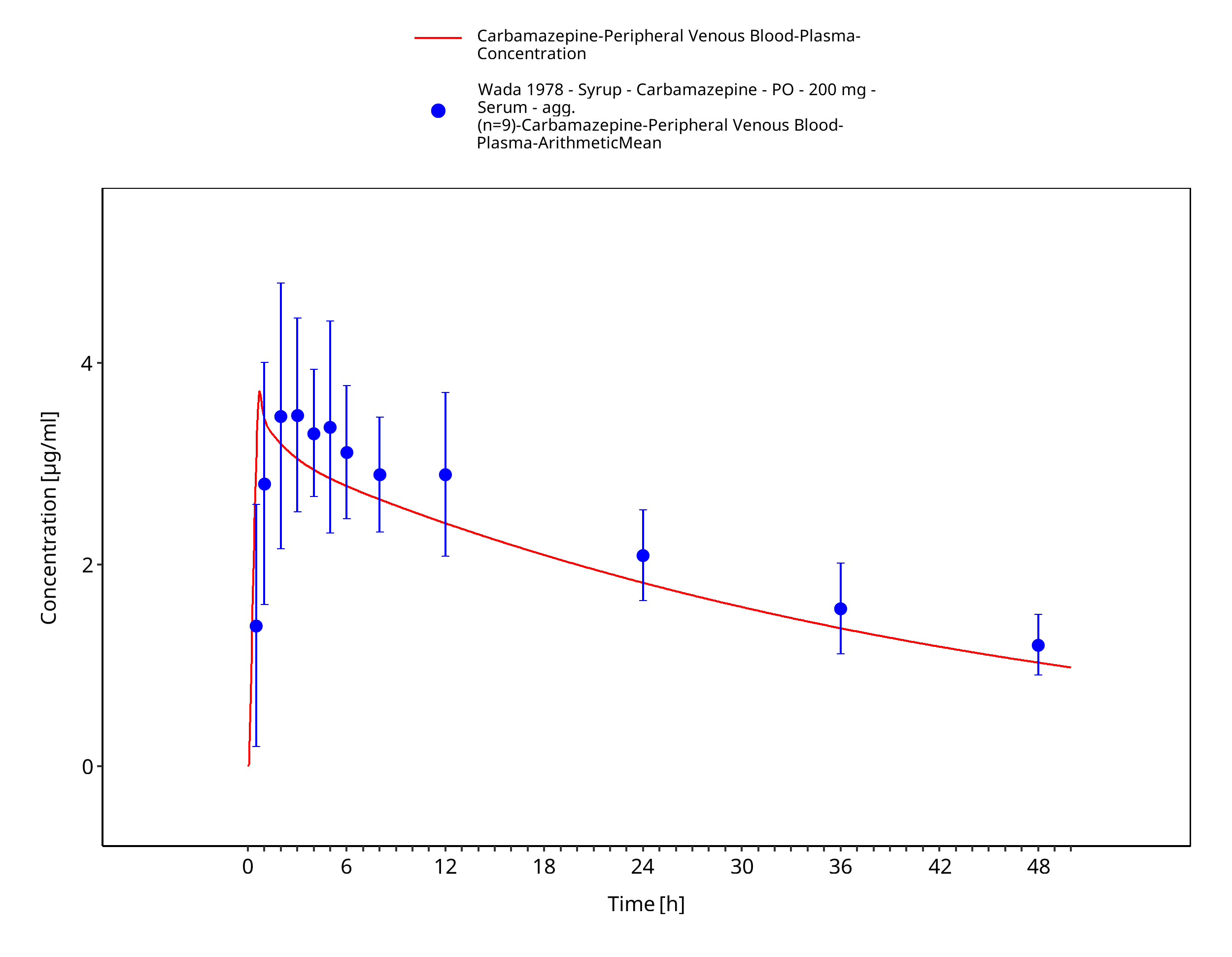
Figure 3-60: Wada1987_200mg_sd_syrup_plasma
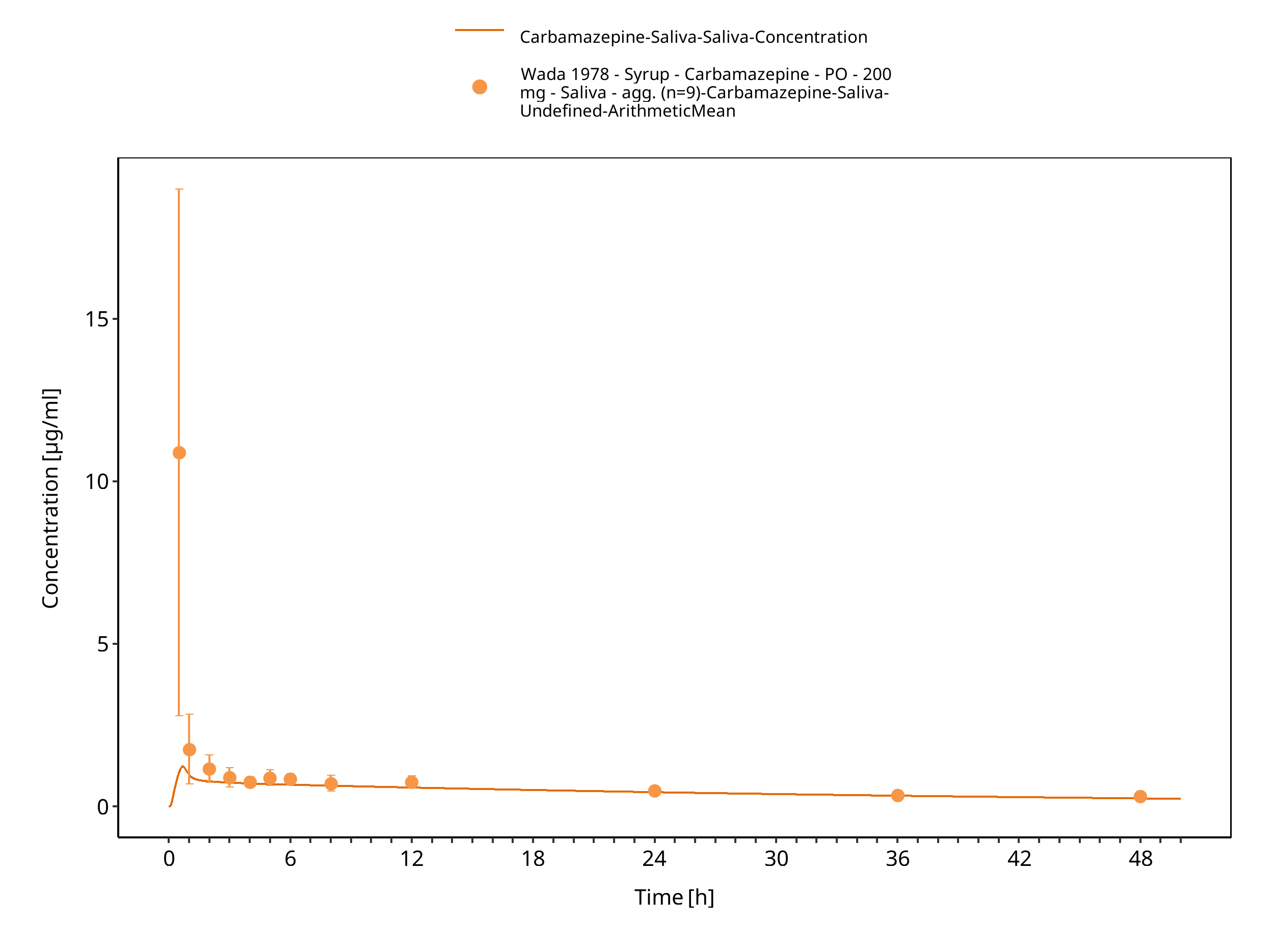
Figure 3-61: Wada1987_200mg_sd_syrup_saliva
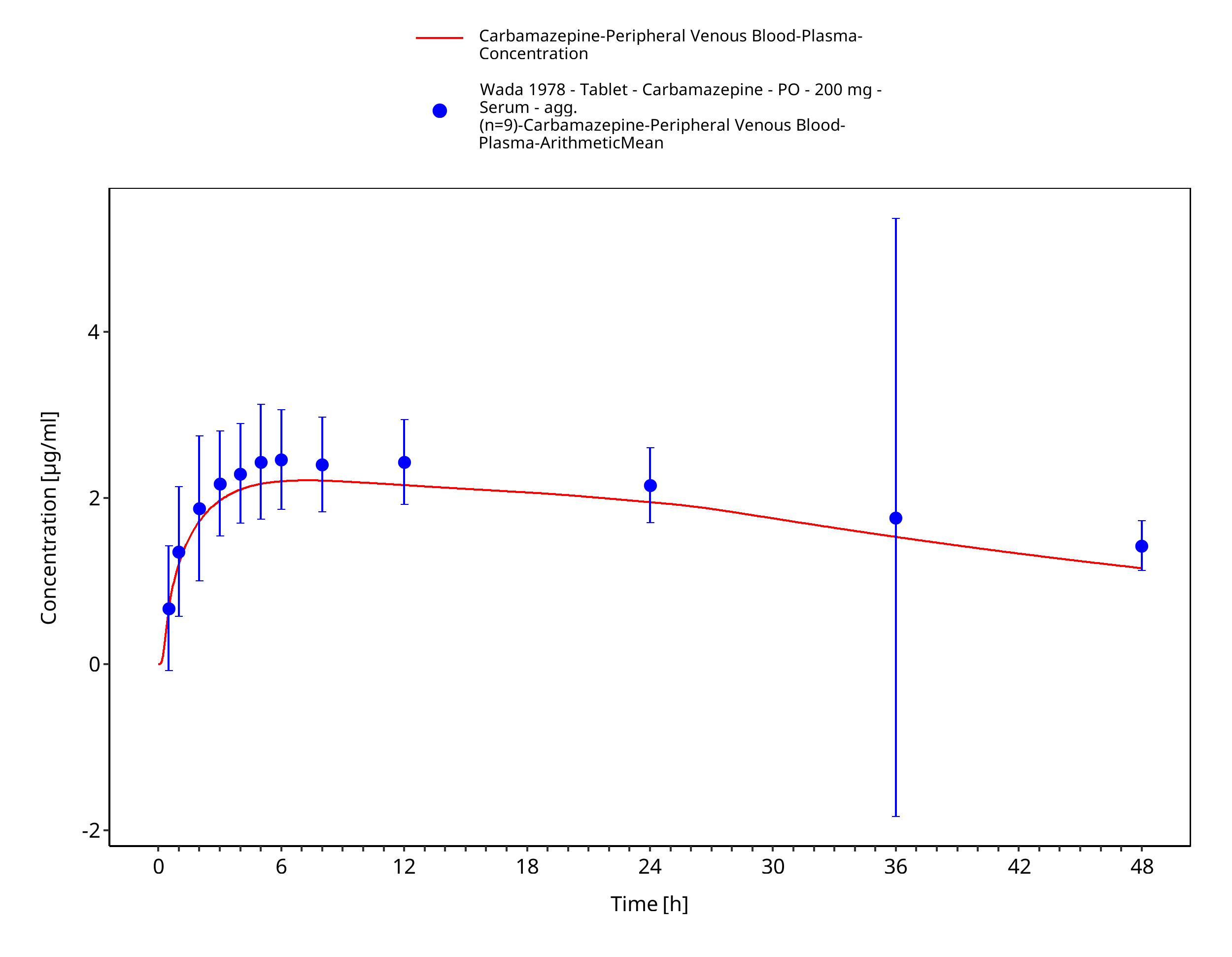
Figure 3-62: Wada1978_200mg_sd_tabIR_plasma
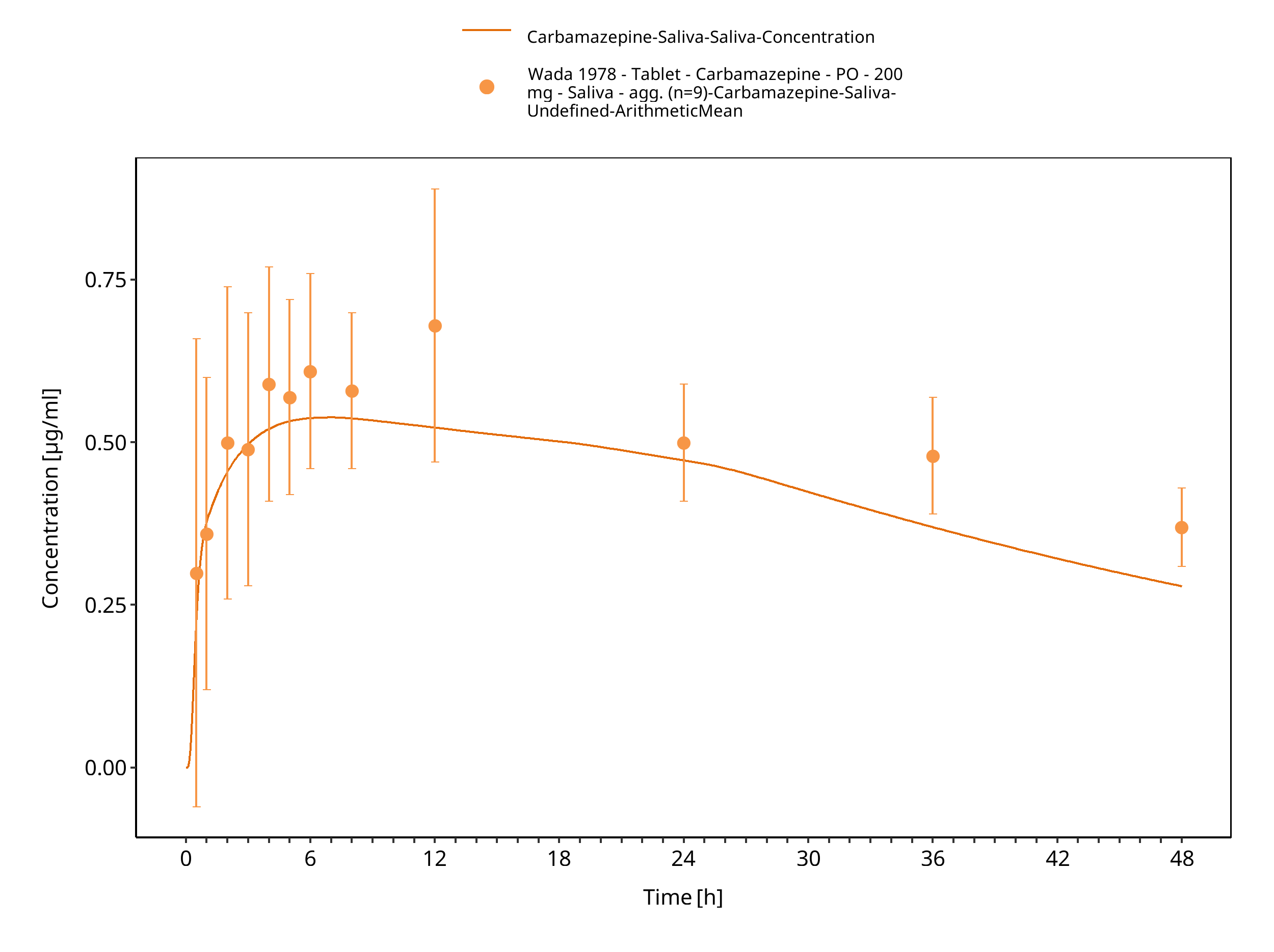
Figure 3-63: Wada1978_200mg_sd_tabIR_plasma 1
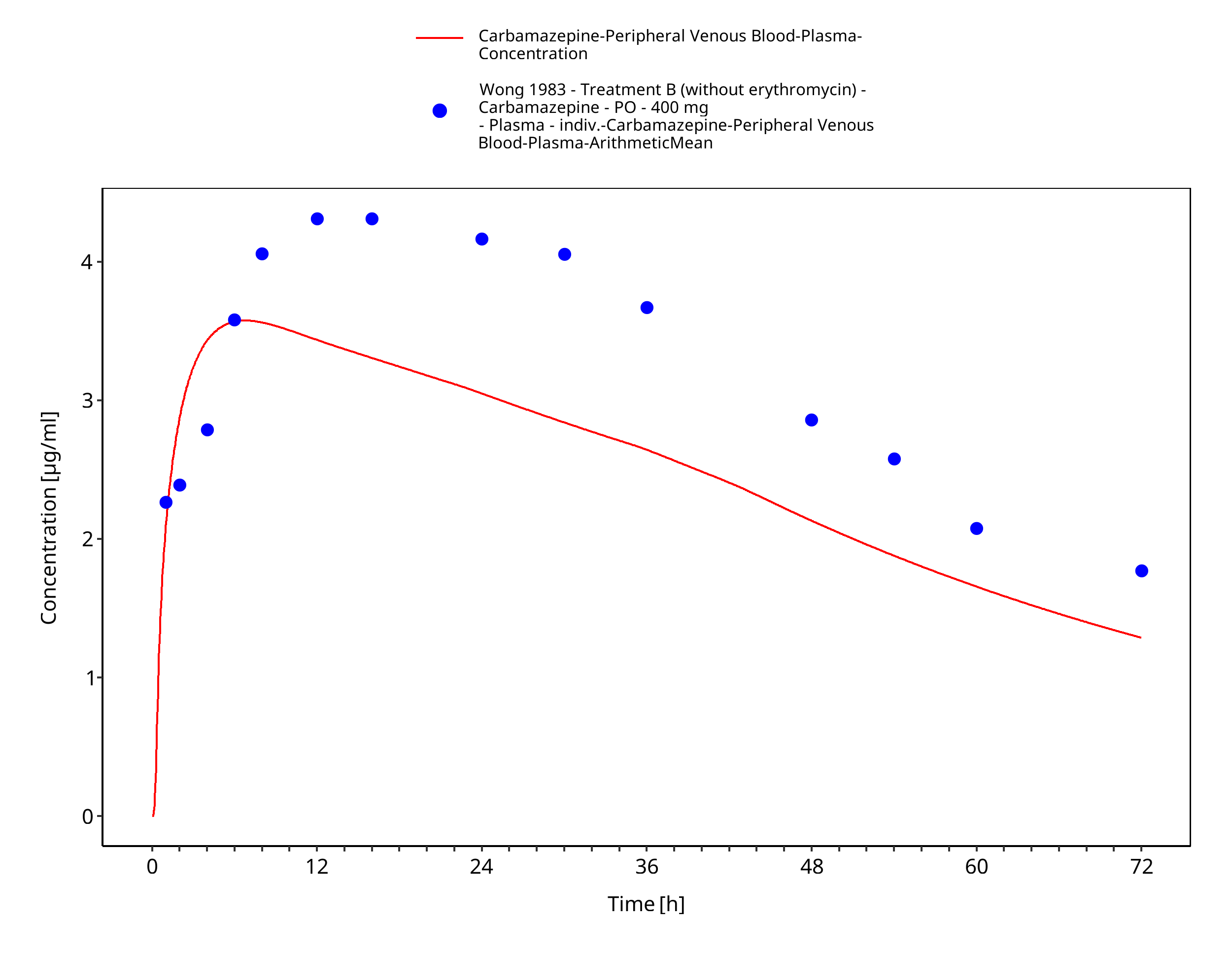
Figure 3-64: Wong1983_control_400mg_sd_tabIR
4 Conclusion¶
The herein presented PBPK model adequately describes the pharmacokinetics of carbamazepine after single and multiple oral administration of various doses to healthy adults.
In conclusion, the presented carbamazepine PBPK model is well-suited to be applied in drug-drug-interaction scenarios.
5 References¶
Achour 2014 Achour, B., Russell, M. R., Barber, J., & Rostami-Hodjegan, A. (2014). Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug metabolism and disposition, 42(4), 500-510.
Almond 2016 Almond, L. M., Mukadam, S., Gardner, I., Okialda, K., Wong, S., Hatley, O., ... & Kenny, J. R. (2016). Prediction of drug-drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metabolism and Disposition, 44(6), 821-832.
Annaert 2010 Annaert, P., Brouwers, J., Bijnens, A., Lammert, F., Tack, J., & Augustijns, P. (2010). Ex vivo permeability experiments in excised rat intestinal tissue and in vitro solubility measurements in aspirated human intestinal fluids support age-dependent oral drug absorption. European journal of pharmaceutical sciences, 39(1-3), 15-22.
Austin 2002 Austin, R. P., Barton, P., Cockroft, S. L., Wenlock, M. C., & Riley, R. J. (2002). The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metabolism and Disposition, 30(12), 1497-1503.
Barzaghi 1987 Barzaghi, N., Gatti, G., Crema, F., Monteleone, M., Amione, C., Leone, L., & Perucca, E. (1987). Inhibition by erythromycin of the conversion of carbamazepine to its active 10, 11‐epoxide metabolite. British journal of clinical pharmacology, 24(6), 836-838.
Bedada 2015 Bedada, S. K., & Nearati, P. (2015). Effect of resveratrol on the pharmacokinetics of carbamazepine in healthy human volunteers. Phytotherapy Research, 29(5), 701-706.
Bedada 2016 Bedada, S. K., Appani, R., & Boga, P. K. (2017). Effect of piperine on the metabolism and pharmacokinetics of carbamazepine in healthy volunteers. Drug research, 67(01), 46-51.
Bernus 1994 Bernus, I., Dickinson, R. G., Hooper, W. D., & Eadie, M. J. (1994). Early stage autoinduction of carbamazepine metabolism in humans. European journal of clinical pharmacology, 47(4), 355-360.
Bianchetti 1987 Bianchetti, G., Padovani, P., Thenot, J. P., Thiercelin, J. F., & Morselli, P. L. (1987). Pharmacokinetic interactions of progabide with other antiepileptic drugs. Epilepsia, 28(1), 68-73.
Burstein 2000 Burstein, A. H., Horton, R. L., Dunn, T., Alfaro, R. M., Piscitelli, S. C., & Theodore, W. (2000). Lack of effect of St John's Wort on carbamazepine pharmacokinetics in healthy volunteers. Clinical Pharmacology & Therapeutics, 68(6), 605-612.
Caraco 1995 Caraco, Y., Zylber-Katz, E., Berry, E. M., & Levy, M. (1995). Carbamazepine pharmacokinetics in obese and lean subjects. Annals of Pharmacotherapy, 29(9), 843-847.
Cawello 2010 Cawello, W., Nickel, B., & Eggert‐Formella, A. (2010). No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. The Journal of Clinical Pharmacology, 50(4), 459-471.
Cazali 2003 Cazali, N., Tran, A., Treluyer, J. M., Rey, E., d’Athis, P., Vincent, J., & Pons, G. (2003). Inhibitory effect of stiripentol on carbamazepine and saquinavir metabolism in human. British journal of clinical pharmacology, 56(5), 526-536.
Clarysse 2011 Clarysse, S., Brouwers, J., Tack, J., Annaert, P., & Augustijns, P. (2011). Intestinal drug solubility estimation based on simulated intestinal fluids: comparison with solubility in human intestinal fluids. European journal of pharmaceutical sciences, 43(4), 260-269.
Cotter 1977 Cotter, L. M., Eadie, M. J., Hooper, W. D., Lander, C. M., Smith, G. A., & Tyrer, J. H. (1977). The pharmacokinetics of carbamazepine. European journal of clinical pharmacology, 12(6), 451-456.
Dalton 1985a Dalton, M. J., Powell, J. R., & Messenheimer Jr, J. A. (1985). The Influence of Cimetidine on Single‐Dose Carbamazepine Pharmacokinetics. Epilepsia, 26(2), 127-130.
Dalton 1985b Dalton, M. J., Powell, J. R., Messenheimer Jr, J. A., Nazario, M., & Mallet, L. (1985). Ranitidine Does Not Alter Single-Dose Carbamazepin Pharmacokinetics in Healthy Adults. Drug intelligence & clinical pharmacy, 19(12), 941-944.
Di Salle 1974 Di Salle, E., Pacifici, G. M., & Morselli, P. L. (1974). Studies on plasma protein binding of carbamazepine. Pharmacological research communications, 6(2), 193-202.
Drugbank DB00564. URL: https://www.drugbank.ca/drugs/DB00564, accessed on 12-14-2020.
Drugbank DBMET00291. URL: https://www.drugbank.ca/metabolites/DBMET00291, accessed on 12-16-2020.
Egnell 2003 Egnell, A. C., Houston, B., & Boyer, S. (2003). In vivo CYP3A4 heteroactivation is a possible mechanism for the drug interaction between felbamate and carbamazepine. Journal of Pharmacology and Experimental Therapeutics, 305(3), 1251-1262.
Eichelbaum 1985 Eichelbaum, M., Tomson, T., Tybring, G., & Bertilsson, L. (1985). Carbamazepine metabolism in man. Clinical pharmacokinetics, 10(1), 80-90.
Elqidra 2004 Elqidra, R., Ünlü, N., Capan, Y., Sahin, G., Dalkara, T., & Hincal, A. A. (2004). Effect of polymorphism on in vitro-in vivo properties of carbamazepine conventional tablets. Journal of Drug Delivery Science and Technology, 14(2), 147-153.
European Patent Application EP 1044681 A2 European Patent Application 2000, EP 1044681 A2, Application no. 00650026.8. URL: https://patentimages.storage.googleapis.com/0c/45/b7/d2be4fa9d24371/EP1044681A2.pdf, accessed on 12-01-2022.
Faucette 2004 Faucette, S. R., Wang, H., Hamilton, G. A., Jolley, S. L., Gilbert, D., Lindley, C., ... & LeCluyse, E. L. (2004). Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metabolism and Disposition, 32(3), 348-358.
Faucette 2007 Faucette, S. R., Zhang, T. C., Moore, R., Sueyoshi, T., Omiecinski, C. J., LeCluyse, E. L., ... & Wang, H. (2007). Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. Journal of Pharmacology and Experimental Therapeutics, 320(1), 72-80.
Fenet 2012 Fenet, H., Mathieu, O., Mahjoub, O., Li, Z., Hillaire-Buys, D., Casellas, C., & Gomez, E. (2012). Carbamazepine, carbamazepine epoxide and dihydroxycarbamazepine sorption to soil and occurrence in a wastewater reuse site in Tunisia. Chemosphere, 88(1), 49-54.
Fuhr 2021 Fuhr, L. M., Marok, F. Z., Hanke, N., Selzer, D., & Lehr, T. (2021). Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics, 13(2), 270.
Gérardin 1976 Gérardin, A. P., Abadie, F. V., Campestrini, J. A., & Theobald, W. (1976). Pharmacokinetics of carbamazepine in normal humans after single and repeated oral doses. Journal of pharmacokinetics and biopharmaceutics, 4(6), 521-535.
Gérardin 1990 Gérardin, A., Dubois, J. P., Moppert, J., & Geller, L. (1990). Absolute bioavailability of carbamazepine after oral administration of a 2% syrup. Epilepsia, 31(3), 334-338.
Hooper 1975 Hooper, W. D., Dubetz, D. K., Bochner, F., Cotter, L. M., Smith, G. A., Eadie, M. J., & Tyrer, J. H. (1975). Plasma protein binding of carbamazepine. Clinical Pharmacology & Therapeutics, 17(4), 433-440.
Huang 2004 Huang, W., Lin, Y. S., McConn, D. J., Calamia, J. C., Totah, R. A., Isoherranen, N., ... & Thummel, K. E. (2004). Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug metabolism and disposition, 32(12), 1434-1445.
Ji 2008 Ji, P., Damle, B., Xie, J., Unger, S. E., Grasela, D. M., & Kaul, S. (2008). Pharmacokinetic interaction between efavirenz and carbamazepine after multiple‐dose administration in healthy subjects. The Journal of Clinical Pharmacology, 48(8), 948-956.
Kayali 1994 Kayali, A., Tuglular, I., & Ertas, M. (1994). Pharmacokinetics of carbamazepine Part I: a new bioequivalency parameter based on a relative bioavailability trial. European journal of drug metabolism and pharmacokinetics, 19(4), 319-325.
Kerr 1994 Kerr, B. M., Thummel, K. E., Wurden, C. J., Klein, S. M., Kroetz, D. L., Gonzalez, F. J., & Levy, R. (1994). Human liver carbamazepine metabolism: role of CYP3A4 and CYP2C8 in 10, 11-epoxide formation. Biochemical pharmacology, 47(11), 1969-1979.
Kim 2005 Kim, K. A., Oh, S. O., Park, P. W., & Park, J. Y. (2005). Effect of probenecid on the pharmacokinetics of carbamazepine in healthy subjects. European journal of clinical pharmacology, 61(4), 275-280.
Kovacević 2009 Kovacević, I., Parojcic, J., Homsek, I., Tubic-Grozdanis, M., & Langguth, P. (2009). Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Molecular pharmaceutics, 6(1), 40-47.
Kuepfer 2016 Kuepfer, L., Niederalt, C., Wendl, T., Schlender, J. F., Willmann, S., Lippert, J., ... & Teutonico, D. (2016). Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT: pharmacometrics & systems pharmacology, 5(10), 516-531.
Lertratanangkoon 1982 Lertratanangkoon, K., & Horning, M. G. (1982). Metabolism of carbamazepine. Drug Metabolism and Disposition, 10(1), 1-10.
Lennernäs 2007 Lennernäs, H. (2007). Intestinal permeability and its relevance for absorption and elimination. Xenobiotica, 37(10-11), 1015-1051.
Levy 1975 Levy, R. H., Pitlick, W. H., Troupin, A. S., Green, J. R., & Neal, J. M. (1975). Pharmacokinetics of carbamazepine in normal man. Clinical Pharmacology & Therapeutics, 17(6), 657-668.
McLean 2001 McLean, A., Browne, S., Zhang, Y., Slaughter, E., Halstenson, C., & Couch, R. (2001). The influence of food on the bioavailability of a twice‐daily controlled release carbamazepine formulation. The Journal of Clinical Pharmacology, 41(2), 183-186.
Meyer 1992 Meyer, M. C., Straughn, A. B., Jarvi, E. J., Wood, G. C., Pelsor, F. R., & Shah, V. P. (1992). The bioinequivalence of carbamazepine tablets with a history of clinical failures. Pharmaceutical Research, 9(12), 1612-1616.
Meyer 1998 Meyer, M. C., Straughn, A. B., Mhatre, R. M., Shah, V. P., Williams, R. L., & Lesko, L. J. (1998). The relative bioavailability and in vivo-in vitro correlations for four marketed carbamazepine tablets. Pharmaceutical research, 15(11), 1787-1791.
Meyer 2012 Meyer, M., Schneckener, S., Ludewig, B., Kuepfer, L., & Lippert, J. (2012). Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metabolism and Disposition, 40(5), 892-901.
Miles 1989 Miles, M. V., & Tennison, M. B. (1989). Erythromycin effects on multiple-dose carbamazepine kinetics. Therapeutic drug monitoring, 11(1), 47-52.
Møller 2001 Møller, S. E., Larsen, F., Khan, A. Z., & Rolan, P. E. (2001). Lack of effect of citalopram on the steady-state pharmacokinetics of carbamazepine in healthy male subjects. Journal of clinical psychopharmacology, 21(5), 493-499.
Morselli 1975 Morselli, P. L., Gerna, M., De Maio, D., Zanda, G., Viani, F., & Garattini, S. (1975). Pharmacokinetic studies on carbamazepine in volunteers and in epileptic patients. In: Clinical pharmacology of anti-epileptic drugs (pp. 166-180). Springer, Berlin, Heidelberg.
Nishimura 2003 Nishimura, M., Yaguti, H., Yoshitsugu, H., Naito, S., & Satoh, T. (2003). Tissue Distribution of mRNA Expression of Human Cytochrome P450 Isoforms Assessed by High-Sensitivity Real-Time Reverse Transcription PCR. Yakugaku zasshi, 123(5), 369-375.
Pearce 2002 Pearce, R. E., Vakkalagadda, G. R., & Leeder, J. S. (2002). Pathways of carbamazepine bioactivation in vitro I. Characterization of human cytochromes P450 responsible for the formation of 2- and 3-hydroxylated metabolites. Drug metabolism and disposition, 30(11), 1170-1179.
Pelkonen 2001 Pelkonen, O., Myllynen, P., Taavitsainen, P., Boobis, A. R., Watts, P., Lake, B. G., ... & Lewis, D. F. V. (2001). Carbamazepine: a 'blind' assessment of CYP-associated metabolism and interactions in human liver-derived in vitro systems. Xenobiotica, 31(6), 321-343.
PK-Sim Ontogeny Database Version 7.3 URL: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/38cf71b384cfc25cfa0ce4d2f3addfd32757e13b/PK-Sim%20Ontogeny%20Database%20Version%207.3.pdf, accessed on 12-01-2022.
Pynnönen 1977 Pynnönen, S. (1977). The pharmacokinetics of carbamazepine in plasma and saliva of man. Acta pharmacologica et toxicologica, 41(5), 465-471.
Rawlins 1975 Rawlins, M. D., Collste, P., Bertilsson, L., & Palmer, L. (1975). Distribution and elimination kinetics of carbamazepine in man. European journal of clinical pharmacology, 8(2), 91-96.
Rodrigues 1999 Rodrigues, A. D. (1999). Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochemical pharmacology, 57(5), 465-480.
Saint-Salvi 1987 Saint-Salvi, B., Tremblay, D., Surjus, A., & Lefebvre, M. A. (1987). A Study of the Interaction of Roxithromycin with Theophylline and Carbarmazepine. Journal of Antimicrobial Chemotherapy, 20(suppl_B), 121-129.
Sakamoto 2013 Sakamoto, M., Itoh, T., & Fujiwara, R. (2013). Prediction of in vivo carbamazepine 10, 11-epoxidation from in vitro metabolic studies with human liver microsomes: importance of its sigmoidal kinetics. Biological and Pharmaceutical Bulletin, 36(12), 1959-1963.
Söderlind 2010 Söderlind, E., Karlsson, E., Carlsson, A., Kong, R., Lenz, A., Lindborg, S., & Sheng, J. J. (2010). Simulating fasted human intestinal fluids: understanding the roles of lecithin and bile acids. Molecular pharmaceutics, 7(5), 1498-1507.
Staines 2004 Staines, A. G., Coughtrie, M. W., & Burchell, B. (2004). N-glucuronidation of carbamazepine in human tissues is mediated by UGT2B7. Journal of Pharmacology and Experimental Therapeutics, 311(3), 1131-1137.
Stevens 1998 Stevens, R. E., Limsakun, T., Evans, G., & Mason, J. D. H. (1998). Controlled, multidose, pharmacokinetic evaluation of two extended‐release carbamazepine formulations (Carbatrol and Tegretol‐XR). Journal of pharmaceutical sciences, 87(12), 1531-1534.
Strandjord 1975 Strandjord, R. E., & Johannessen, S. I. (1975). A preliminary study of serum carbamazepine levels in healthy subjects and in patients with epilepsy. In: Clinical pharmacology of anti-epileptic drugs (pp. 181-188). Springer, Berlin, Heidelberg.
Sumi 1987 Sumi, M., Watari, N., Umezawa, O., & Kaneniwa, N. (1987). Pharmacokinetic study of carbamazepine and its epoxide metabolite in humans. Journal of pharmacobio-dynamics, 10(11), 652-661.
Terhaag 1978 Terhaag, B., Richter, K., & Diettrich, H. (1978). Concentration behavior of carbamazepine in bile and plasma of man. International journal of clinical pharmacology and biopharmacy, 16(12), 607-609.
Tomaszewska 2013 Tomaszewska, I. (2013). In vitro and Physiologically Based Pharmacokinetic models for pharmaceutical cocrystals. Doctoral dissertation, University of Bath. https://purehost.bath.ac.uk/ws/portalfiles/portal/187934939/UnivBath_PhD_2013_I_Tomaszewska.pdf, accessed on 12-01-2022.
Tomson 1983 Tomson, T., Tybring, G., & Bertilsson, L. (1983). Single‐dose kinetics and metabolism of carbamazepine‐10, 11‐epoxide. Clinical Pharmacology & Therapeutics, 33(1), 58-65.
US Patent Application - US 2009/0169619 A1. United States Patent Application 2009, Publication no.: US 2009/0169619 A1. https://patentimages.storage.googleapis.com/66/d4/30/f3588f44ab2b6f/US20090169619A1.pdf, accessed on 12-01-2022.
US Patent Application - US 2014/0302138 A1. United States Patent Application 2014, Publication no.: US 2014/0302138 A1. https://patentimages.storage.googleapis.com/57/d9/18/0d8cbfa046681d/US20140302138A1.pdf, accessed on 12-01-2022.
Vinçon 1987 Vinçon, G., Albin, H., Demotes-Mainard, F., Guyot, M., Bistue, C., & Loiseau, P. (1987). Effects of josamycin on carbamazepine kinetics. European journal of clinical pharmacology, 32(3), 321-323.
Wada 1978 Wada, J. A., Troupin, A. S., Friel, P., Remick, R., Leal, K., & Pearmain, J. (1978). Pharmacokinetic comparison of tablet and suspension dosage forms of carbamazepine. Epilepsia, 19(3), 251-255.
Williamson 2016 Williamson, B., Lorbeer, M., Mitchell, M. D., Brayman, T. G., & Riley, R. J. (2016). Evaluation of a novel PXR‐knockout in HepaRG™ cells. Pharmacology research & perspectives, 4(5), e00264.
Willmann 2007 Willmann, S., Höhn, K., Edginton, A., Sevestre, M., Solodenko, J., Weiss, W., ... & Schmitt, W. (2007). Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. Journal of pharmacokinetics and pharmacodynamics, 34(3), 401-431.
Wong 1983 Wong, Y. Y., Ludden, T. M., & Bell, R. D. (1983). Effect of erythromycin on carbamazepine kinetics. Clinical Pharmacology & Therapeutics, 33(4), 460-464.
Related Models¶
Explore other Anticonvulsants models in this library:
- S-Mephenytoin - PBPK model and evaluation report