Population simulations
Population simulations can be easily performed in R by combining the simulation loaded from a *.pkml file with the population information created in PK-Sim and exported to CSV format (for details, please refer to OSPS online documentation) or created directly in R (see Creating populations).
Loading population file
The method loadPopulation creates an object of the
Population class that can be passed to the
runSimulations() method (see Running simulations and retrieving the
results).
library(ospsuite)
# Load population information from csv
popFilePath <- system.file("extdata", "pop.csv", package = "ospsuite")
myPopulation <- loadPopulation(csvPopulationFile = popFilePath)
print(myPopulation)
#> <Population>
#> • Number of Individuals: 10Creating populations
Similar to creating individual parameter sets (see Creating individuals), a population is
created from population characteristics created by calling the
method createPopulationCharacteristics(). To see the list
of available values for the arguments species and
population (only for human), use the enums
Species and HumanPopulation, respectively. The
returned object of type PopulationCharacteristics is then
passed to the function createPopulation to generate a set
of parameter values. The algorithm behind is the same used in PK-Sim
when creating an population. Molecule ontogenies can be added as
described in the vignette Creating
individuals.
library(ospsuite)
# If no unit is specified, the default units are used. For "height" it is "dm", for "weight" it is "kg", for "age" it is "year(s)".
populationCharacteristics <- createPopulationCharacteristics(
species = Species$Human,
population = HumanPopulation$Asian_Tanaka_1996,
numberOfIndividuals = 50,
proportionOfFemales = 50,
weightMin = 30,
weightMax = 98,
weightUnit = "kg",
heightMin = NULL,
heightMax = NULL,
ageMin = 0,
ageMax = 80,
ageUnit = "year(s)"
)
print(populationCharacteristics)
#> <PopulationCharacteristics>
#> • Species: Human
#> • Population: Asian_Tanaka_1996
#> • Number of individuals: 50
#> • Proportion of females: 50
#> • Age: [0.00 year(s)..80.00 year(s)]
#> • Gestational age: ]-Inf..+Inf[
#> • Weight: [30.00 kg..98.00 kg]
#> • Height: ]-Inf..+Inf[
#> • BMI: ]-Inf..+Inf[
# Create population from population characteristics
result <- createPopulation(populationCharacteristics = populationCharacteristics)
myPopulation <- result$population
print(myPopulation)
#> <Population>
#> • Number of Individuals: 50Running population simulation
To run a population simulation, the Population object
created by the createPopulation method must be passed to
the runSimulation() method:
library(ospsuite)
# Load simulation
simFilePath <- system.file("extdata", "Aciclovir.pkml", package = "ospsuite")
sim <- loadSimulation(simFilePath)
# Run population simulation
simulationResults <- runSimulations(simulations = sim, population = myPopulation)[[1]]
print(simulationResults)
#> <SimulationResults>
#> • Number of individuals: 50
#> For paths:
#> • Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)
#> • Organism|VenousBlood|Plasma|Aciclovir|Plasma UnboundPopulation simulations are run in parallel on multi-core machines - one core simulates a subset of all individuals defined in the population. By default, the number of cores used equals the maximal number of logical cores available minus one.
The user can change the default behavior by providing custom
SimulationRunOptions().
# Load simulation
simFilePath <- system.file("extdata", "Aciclovir.pkml", package = "ospsuite")
sim <- loadSimulation(simFilePath)
# Create a SimulationRunOptions object
simRunOptions <- SimulationRunOptions$new()
print(simRunOptions)
#> <SimulationRunOptions>
#> • numberOfCores: 3
#> • checkForNegativeValues: TRUE
#> • showProgress: FALSE
# Change the maximal number of cores to use and show a progress bar during simulation
simRunOptions$numberOfCores <- 3
simRunOptions$showProgress <- TRUE
# Run population simulation with custom options
populationResults <- runSimulations(simulations = sim, population = myPopulation, simulationRunOptions = simRunOptions)[[1]]
print(populationResults)
#> <SimulationResults>
#> • Number of individuals: 50
#> For paths:
#> • Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)
#> • Organism|VenousBlood|Plasma|Aciclovir|Plasma UnboundSimulated time-value pairs for a specific output from the
SimulationResults-object returned by the
runSimulation method can be accessed with the method
getOutputValues. The user can provide either the path(s) of
the output (which can be a molecule, a parameter, or an observer), or
the object(s) of the type Molecule, Parameter,
or Quantity (for observers) with the argument
quantitiesOrPaths. If no output is specified, all outputs
available in the simulation results are returned.
The paths of all available outputs can be accessed via
populationResults$allQuantityPaths
#> [1] "Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)"
#> [2] "Organism|VenousBlood|Plasma|Aciclovir|Plasma Unbound"getOutputValues() returns a list with two entries:
data and metadata:
-
datais a dataframe with two predefined columns (IndividualId and Time) as well as one column for each requested outputIndividualId-
Timea vector with simulated time values (in minutes, equal for all outputs) - a vector with simulated entries for each output requested.
The values of IndividualId, Time, and the
simulated outputs, are appended for each simulated individual. Note that
this results in non-monotonously increasing column
Time.
# Get simulated results by path
resultsPath <- populationResults$allQuantityPaths[[1]]
print(resultsPath)
#> [1] "Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)"
resultsData <- getOutputValues(populationResults, quantitiesOrPaths = resultsPath)
resultsTime <- resultsData$data$Time
resultsValues <- resultsData$data$`Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)`
plot(resultsTime, resultsValues, type = "l")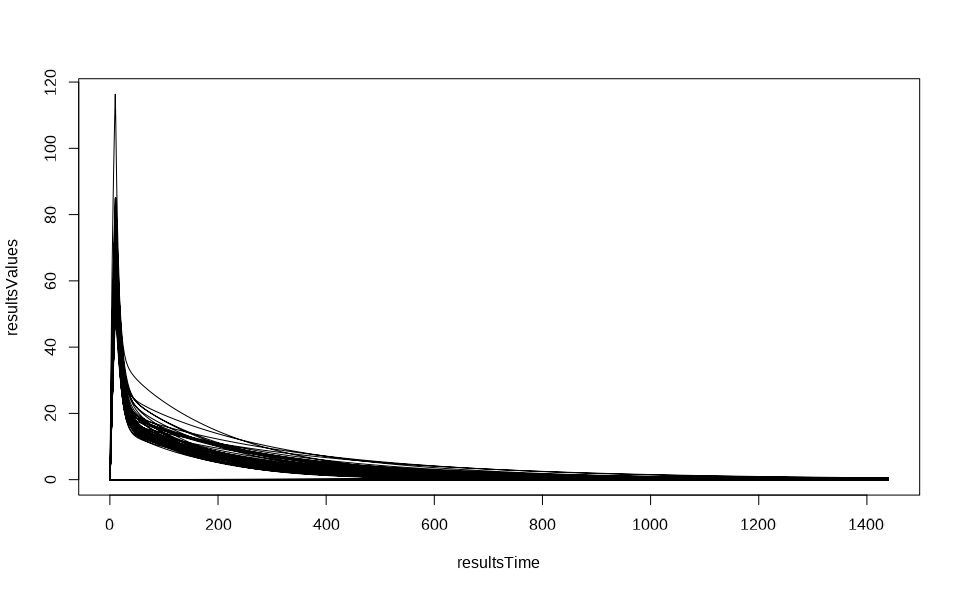
To get the results for a specific individual or a set of individuals,
the argument individualIds of the method
getOutputValues() can be specified:
# Get simulated results by path
resultsPath <- populationResults$allQuantityPaths[[1]]
print(resultsPath)
#> [1] "Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)"
# Get only the results for individuals with IDs 1 and 2
resultsData <- getOutputValues(populationResults, quantitiesOrPaths = resultsPath, individualIds = c(1, 2))
resultsTime <- resultsData$data$Time
resultsValues <- resultsData$data$`Organism|PeripheralVenousBlood|Aciclovir|Plasma (Peripheral Venous Blood)`
plot(resultsTime, resultsValues, type = "l")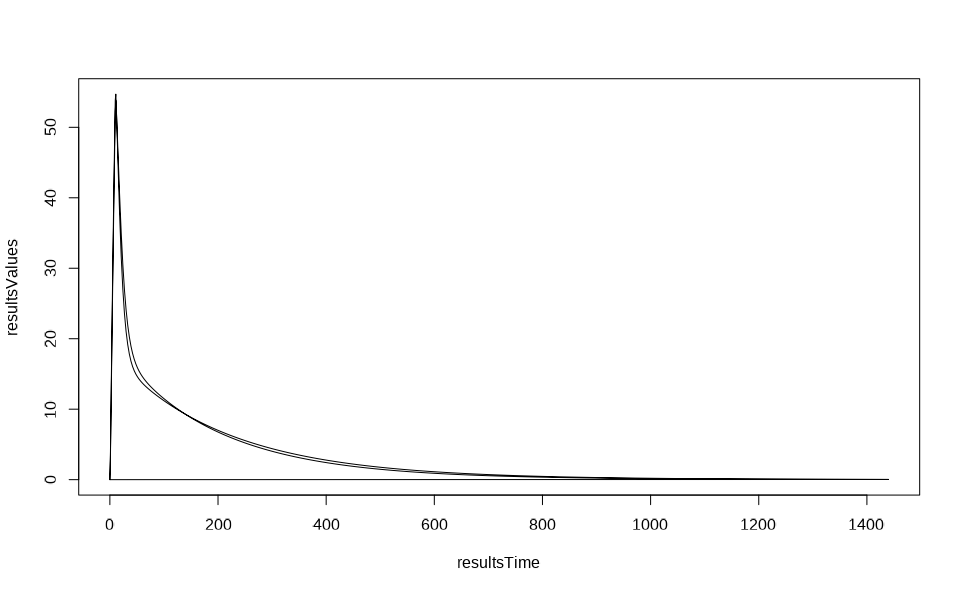
For more information about running simulations, please refer to Running simulations and retrieving the results.